



Shrinidh Joshi, especialista em dispositivos médicos on Kolabtree, provides a comprehensive guide to dispositivo médico design, design controls, validation & verification, regulatory requirements and risk management.
No artigo anterior, we took a look at the overview of the dispositivo médico development process from the ideation to the discovery phase. In this article, we will focus more on medical device design, design controls, and compliance.
Projeto de dispositivos médicos: Regulamentos IEC & ISO e conformidade
Até agora você sabe que para entrar no mercado seu dispositivo médico precisa qualificar certos requisitos e padrões regulamentares. Os padrões de produtos médicos, como a Comissão Eletrotécnica Internacional (IEC) ou a Organização Internacional de Normalização (ISO) permitem que fabricantes de produtos médicos, projetistas, laboratórios e todos os outros prestadores de serviços de desenvolvimento de produtos médicos, como o CDMO, inspecionem, avaliem e mantenham seus dispositivos e equipamentos de acordo com certos padrões de qualidade e usabilidade.
A (IEC publicou a primeira norma de dispositivos médicos de seu tipo em 1970, IEC 60601-1. IEC 60601-1, Equipamento elétrico médico - Parte 1: Esta é a norma reconhecida internacionalmente que trata de requisitos gerais para equipamentos e dispositivos elétricos médicos cobrindo normas para segurança básica e desempenho essencial. [4].
O documento IEC 60601-1 foi revisado periodicamente para ficar em alinhamento com os mais recentes desenvolvimentos médicos e avanços tecnológicos no campo de dispositivos médicos. A mudança mais recente foi realizada em 2012 (Emenda 1 à IEC 60601-1). Estas normas revisadas contêm os requisitos para consideração do fator humano, avaliação essencial de desempenho de dispositivos médicos, usabilidade e comandos. Ela também inclui software como um dispositivo médico e especifica sua adoção de um ciclo de vida de desenvolvimento formal. Também incluídos no escopo da IEC 60601-1 revisada estão especificações técnicas mais novas e revisadas para riscos (elétricos e mecânicos), requisitos de rotulagem de dispositivos médicos (incluindo novas normas de rotulagem) e documentação.
Projeto de dispositivos médicos: Normas ISO
A Organização Internacional de Padronização também tem especificações para normas de dispositivos médicos. ISO 13485 e ISO 14971 são padrões amplamente utilizados em todo o mundo para a gestão da qualidade de dispositivos médicos. Além dessas normas internacionais, certas normas são específicas para cada região e todas elas são adotadas a partir de normas internacionais com pouca modificação e limitação.
Se uma empresa de produtos médicos estiver fabricando ou vendendo produtos médicos nos EUA, o dispositivo médico será regulamentado pela FDA. O American National Standards Institute (ANSI) é o representante das normas ISO nos EUA.
Há mais duas organizações similares: a Association for the Advancement of Medical Instrumentation (AAMI) e a American Society for Quality (ASQ), que define padrões para os EUA.
Se uma empresa de produtos médicos tivesse projetado um dispositivo considerando as normas ISO, há também a possibilidade de que a FDA não aprove o dispositivo. Como a FDA tem seu próprio conjunto de procedimentos para gerenciamento de risco derivado de normas internacionais e regionais, o que inclui:
- ISO 14971:2007, Medical devices – Application of risk management to medical devices
(padrão internacional).
- ANSI/AAMI/ISO 14971:2007 (R2010), Medical devices - Application of risk management to medical devices (Uma norma regional com adições e modificações da referida norma internacional). [5].
No caso da norma de gestão de qualidade, ela não segue a versão internacional ou regional da norma ISO 13485. Isto porque a FDA tem diretrizes diferentes para o gerenciamento de qualidade em dispositivos médicos para o mercado dos EUA.
Considerando que, se a empresa de produtos médicos está considerando a União Européia, o Comitê Europeu de Normalização (CEN) é a padronização adotada pela ISO e o Comitê Europeu de Normalização Eletrotécnica (CENELEC) é a norma regional inspirada pela IEC.
CEN é um pouco modificado conforme a exigência da ISO e escrito com um prefixo "EN". Por exemplo:
- EN ISO 13485:2012, Dispositivos médicos - Sistemas de gestão de qualidade - Requisitos para fins regulamentares.
- EN ISO 14971:2012, Dispositivos médicos - Aplicação da gestão de risco aos dispositivos médicos
Os membros nacionais adotam estas normas da UE ao mesmo tempo em que acrescentam seu prefixo. Para a Suíça, a Swiss Standards publica normas com "SN" como prefixo, como SN EN ISO 13485:2012 e SN EN ISO 14971:2012.
No caso do Canadá, a Canadian Standards Authority (CSA) é a organização representativa da ISO.
Regulamentos e Controle de Projeto de Dispositivos Médicos
Os fabricantes de dispositivos médicos precisam seguir as diretrizes de Controle de Projeto, pois os órgãos reguladores como FDA, Comissão Européia, Health Canada e outros querem garantir que os dispositivos médicos sejam seguros para os usuários potenciais antes que os fabricantes comecem a comercializar os dispositivos. Como mencionei na seção acima, a FDA não segue a ISO 13485, pois tem requisitos diferentes para o gerenciamento da qualidade. Os controles de projeto são definidos em FDA 21 CFR 820.30 que tem uma intenção semelhante à seção 7.3 Projeto e Desenvolvimento descrita sob as diretrizes da ISO 13485. Além disso, a FDA incorpora as exigências das Boas Práticas de Fabricação Atuais (cGMP) no regulamento do sistema de qualidade para seguir as boas práticas de qualidade para projetos de dispositivos médicos. [6].
O regulamento fornece uma estrutura para implementar o controle do projeto para uma grande variedade de dispositivos. A estrutura proporciona flexibilidade tanto para as exigências regulamentares quanto para o processo interno de projeto e desenvolvimento.
Para implementar com sucesso o controle de projetos de dispositivos médicos, são necessários profissionais com formação técnica e não técnica, como administração de empresas, ciências da vida, engenharia, ciência da computação e artes.
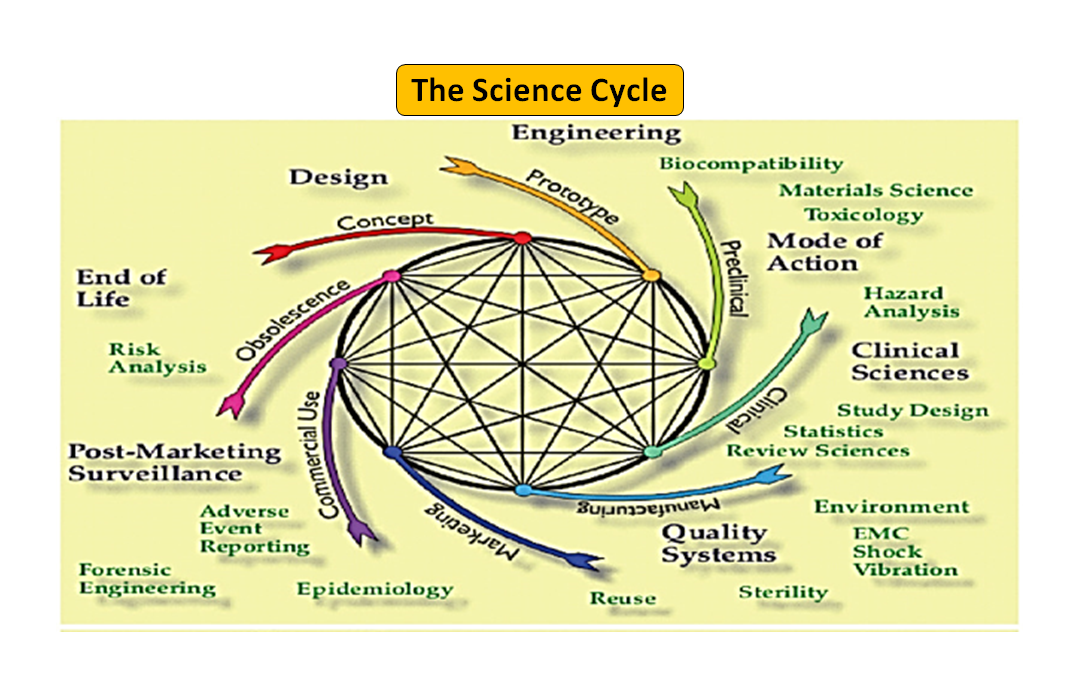
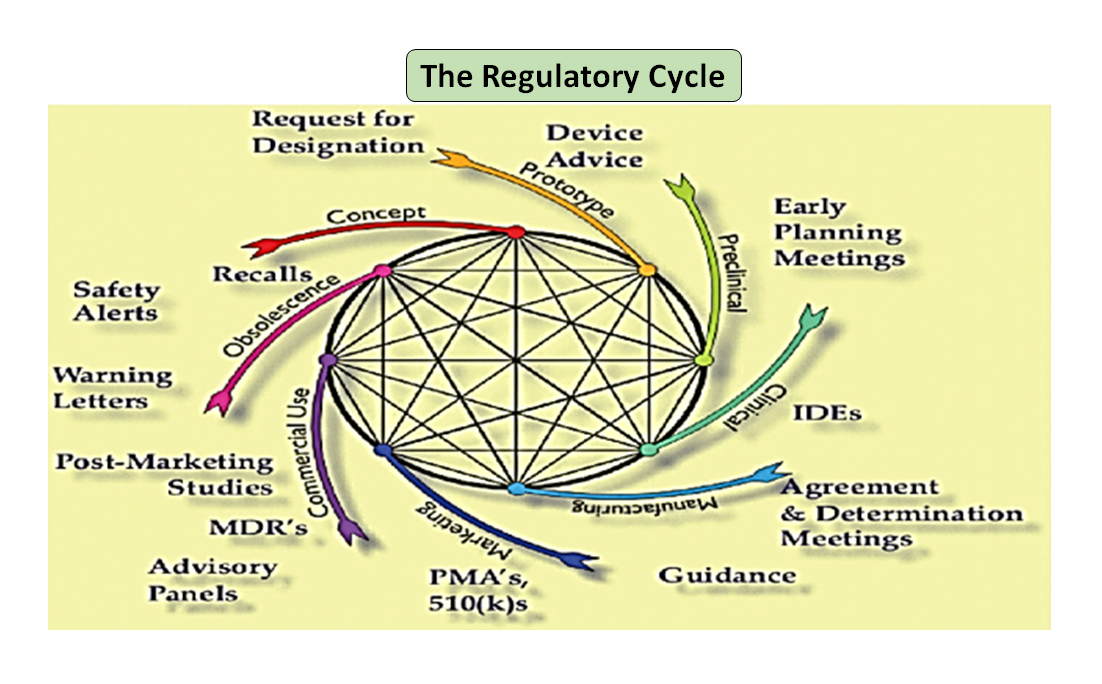
Figura 1: Ciclo de vida total do produto. O ciclo da ciência e o ciclo regulamentar (Adaptado de [7]).
É digno de nota que o ciclo de vida do dispositivo médico desde a inovação até a aprovação e comercialização regulamentar é uma série de etapas interligadas que impulsiona o desenvolvimento do dispositivo (ver Figura 1: Ciclo total do produto). No início, os protótipos projetados por seus engenheiros são testados em bancada para otimizar o projeto, testados quanto à biocompatibilidade, extratáveis, lixiviáveis, flexibilidade ou resistência geral de seu dispositivo. O papel do consultor de regulamentação de sua empresa é navegar pelo banco de dados de regulamentação para sugerir a você um documento de orientação que pode ajudá-lo a determinar se seu produto será ou não regulamentado como dispositivo médico. O uso pretendido de seu dispositivo médico e seu modo de operação ou ação o orientará no projeto do dispositivo e também decidirá sua via regulatória se 510(k), PMA, De Novo, Pre-sub, IDE, HDE, arquivos principais etc.
Como ilustrado na Figura 1, tanto os processos científicos quanto os regulamentares estão interligados ao longo de todo o ciclo de vida do produto. Assim como diferentes partes do ciclo de vida da ciência estão interligadas, a ciência e os requisitos regulamentares estão entrelaçados, cada um informando e determinando o outro. Há uma oportunidade de construir conexões, tanto na FDA como nos fabricantes, de modo que partes do ciclo de vida não correm o risco de serem consideradas isoladamente. Por exemplo, não é raro que uma aplicação pré-comercialização seja revista sem considerar a experiência pós-mercado de produtos similares.
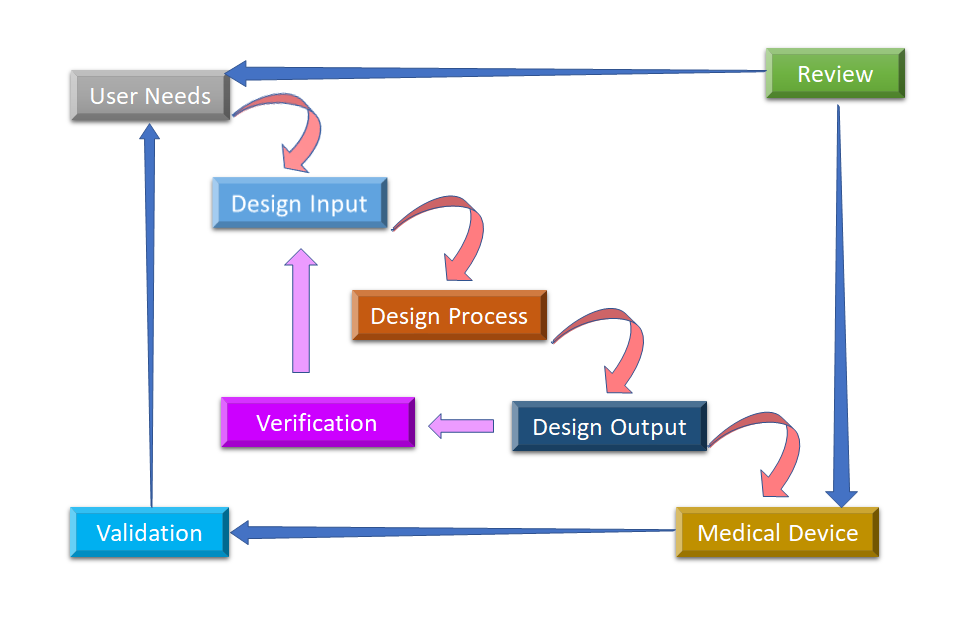
Figura 2: Processo de projeto de fluxo de água para controle de projeto de dispositivos médicos (Adaptado de [8]).
A fase inicial a partir da qual se inicia o Controle de Projeto é o desenvolvimento e aprovação do Design Input, que consiste no projeto do dispositivo e processos de fabricação a serem realizados na fase de produção. O Controle de Projeto é uma abordagem holística e não termina com a transferência do projeto para a fase de produção, uma vez que o projeto é finalizado. Ele também impacta os processos de fabricação de acordo com as mudanças na fase de projeto ou mesmo com o feedback pós-produção. É um processo contínuo para desenvolver um produto que seja utilizável por um usuário e, portanto, para o produto aprimorado, considera mudanças revolucionárias em relação aos padrões de uso, bem como a análise de produtos defeituosos. Você pode ver na Figura 2, como o Controle de Projeto pode ser realizado no processo de projeto da cascata.
- Necessidades do usuário:- Os requisitos são definidos considerando a necessidade do mercado e o dispositivo é projetado para atender a essa necessidade. Após uma série de evoluções, o projeto do dispositivo médico é finalizado e transferido para a produção para fabricação. Há uma necessidade de feedback durante cada etapa deste processo.
- Entrada de projeto: Este é um processo iterativo. Quando uma organização decide atender a uma necessidade específica, eles revisam e testam a aceitabilidade do projeto derivado da necessidade. Nesse ponto, inicia-se o processo iterativo de conversão de requisitos em design de dispositivos.
- Processo de projeto: Essas entradas de projeto são convertidas em saídas de projeto, convertendo essas exigências em especificações de alto nível (que são saídas de projeto).
- Saída de projeto: O processo de verificação confirma se as especificações estão satisfazendo os requisitos ou não. E a saída torna-se a entrada para revisar os requisitos e este processo continua até que a Saída do Projeto esteja alinhada com a Entrada do Projeto.
- Dispositivo Médico: Once the final design is ready, it is transmitted to the production facility for mass manufacturing. Design control regulation mandates Design History File (DHF), which illustrates the linkages and relationships between all the Design Controls and help to trace all changes throughout the entire desenvolvimento de produtos processo.
As empresas de dispositivos médicos podem adotar uma abordagem baseada em papel ou em software, especialmente desenvolvida para o Controle de Projeto; os arquivos de histórico de projeto devem ser rastreáveis, bem como acessíveis a todos os membros da equipe.
O fluxograma abaixo mostra um estudo de caso para o controle do projeto de dispositivos médicos.

Projeto de dispositivos médicos: Por que a Rastreabilidade é Importante
Currently in the realm of the medical devices industry, it is an ideal practice to develop a traceability matrix that can illustrate the links and relations between user needs, design inputs and outputs, design verification and validation. When you are in the early phase for your device development you can maintain the device traceability using a spreadsheet or document version but as you move forward, its good idea to use cloud-based project management and document sharing platforms such as Microsoft Teams, Asana, Trello or whichever platform is suitable for your organization. The goal is as your project progresses you need to find an option which can save time because the old-school method of maintaining a traceability matrix might consume a lot of your time which you should rather be focusing on design verification and validation.
Uma matriz de rastreabilidade do Controle de Projeto é vital para as equipes de desenvolvimento de produtos, e especialmente para os gerentes de projeto, porque a rastreabilidade mostra a relação e as ligações entre todos os Controles de Projeto. Como as necessidades do usuário se relacionam com as entradas do Design? Como as saídas do Design se relacionam com as entradas do Design? Como as Verificações de Projeto se ligam às Entradas e Saídas do Projeto? Como as Validações de Projeto se relacionam com as Necessidades do Usuário? Uma matriz de rastreabilidade é uma ferramenta inestimável para mostrar uma visão de alto nível e o fluxo de desenvolvimento de produtos de dispositivos médicos do início ao fim.
Os desenvolvedores de produtos de melhores práticas têm confiado na rastreabilidade do Design Controls há muitos, muitos anos. E agora ISO 13485:2016 também faz da rastreabilidade uma exigência. Como citado na ISO 13485:2016, 7.1 Planejamento da Realização do Produto, 1. c) verificação, validação, monitoramento, medição, inspeção e teste, manuseio, armazenamento, distribuição e atividades de rastreabilidade específicas para o produto, juntamente com os critérios para aceitação do produto; e 7.3.2 Planejamento do Projeto e Desenvolvimento, 1. e) os métodos para garantir a rastreabilidade dos resultados do projeto e desenvolvimento para as entradas do projeto e desenvolvimento [9].
Projeto de dispositivos médicos: Verificação e Validação
Todo dispositivo médico deve atender aos objetivos de funcionalidade, usabilidade e confiabilidade para obter uma participação bem sucedida no mercado.
Além disso, suas partes interessadas (pacientes, prescritores, reguladores ou usuários finais) também prestarão atenção à segurança e eficácia de seu dispositivo. É muito provável que seu dispositivo seja projetado para atender a uma necessidade não atendida que pode ser crítica para a vida, por exemplo, um ventilador ou um dispositivo de diagnóstico que possa detectar doenças cardíacas. Portanto, o teste iterativo do seu dispositivo com verificação e validação é crucial. Estas duas etapas no processo de projeto visam confirmar que seu dispositivo médico está alinhado com as exigências dos usuários e que está funcionando de acordo com seu uso pretendido. Em termos simples, a verificação e validação do projeto pode assegurar que seu dispositivo está realmente fazendo o que se supõe que esteja fazendo. A verificação e a validação do projeto também são para garantir os requisitos regulamentares, as normas, a qualidade do produto e o processo de fabricação de seu dispositivo médico. A verificação do projeto pode avaliar se seu projeto está em conformidade com as exigências, especificações ou requisitos regulamentares especificados na entrada do projeto. Por outro lado, a validação do projeto é destinada a avaliar se seu dispositivo médico está proporcionando benefícios com base na necessidade dos usuários finais.
Projeto verificação pergunta: "Nós projetamos o dispositivo corretamente?"
Projeto validação pergunta: "Nós projetamos o dispositivo certo?"
Os dispositivos médicos podem consistir de diferentes formas tecnológicas, tamanhos e diferentes níveis de complexidade. A atividade de verificação e validação (V&V) é impulsionada pelo ambiente regulatório e deve seguir padrões internacionais. Atividades de V&V padronizadas podem agilizar o processo de fabricação, assim como melhorar o processo de aprovação. Além disso, testes automatizados, técnicas de diagnóstico e ferramentas de coleta de dados podem aprimorar o processo de V&V. [10].
- Validação do produto vs. Validação do processo
- Projeto do dispositivo médico/validação do produto:- Em conformidade com as necessidades do usuário e do paciente, ou seja, o dispositivo funciona corretamente?
- Validação do processo:- O processo de fabricação atende a especificações pré-determinadas.
Vale lembrar a validação do projeto/validação do produto ≠ validação do processo. As agências reguladoras exigem tanto a validação do projeto/produto quanto a validação do processo individualmente, portanto ambos precisam ser levados em consideração igualmente durante a submissão regulamentar.
Em que fase inicial do processo de desenvolvimento devemos pensar na validação? Uma empresa de dispositivos médicos deve entender que nunca é cedo demais para iniciar o trabalho de validação, uma empresa deve começar a validar mais cedo do que tarde para descobrir que está seguindo o caminho certo e resolver o problema certo.
A validação (também V&V) sendo um processo iterativo consome um bom investimento, quando mal planejado. Uma estratégia de teste fortemente definida pode ajudá-lo a otimizar os custos, bem como o período de teste para preparar o produto para o mercado em tempo hábil.
A complexidade de qualquer estratégia de teste depende das tecnologias a serem utilizadas e dos mercados-alvo geográficos. A estratégia de teste deve cobrir pelo menos seis parâmetros mencionados abaixo:
- Geografias direcionadas e padrões associados;
- Tempo para comercializar;
- Um padrão a ser seguido com uma versão;
- Laboratórios de testes - laboratórios internos ou independentes;
- Definindo a seqüência de testes;
- Apresentando o resultado do teste
Assim, os testes utilizados para o processo de verificação e validação também precisam ser validados. Isto é para garantir que você meça o que precisa medir porque um teste errado produzirá resultados errados de usabilidade e funcionalidade. As empresas de dispositivos médicos precisam de uma V&V eficaz e bem documentada, que esteja em conformidade com os regulamentos associados.
Projeto de dispositivos médicos: Gerenciamento de risco
Estratégia de migração de risco vs. Plano de gerenciamento de risco
Os procedimentos de gerenciamento de risco para dispositivos médicos são aplicados sob normas de conformidade internacionalmente aceitas ISO 14971:2007 Medical Devices - "Application of Risk Management to Medical Devices”. Além disso, as políticas de gerenciamento de risco precisam ser incorporadas em todas as etapas de projeto e desenvolvimento de dispositivos médicos e também devem ser associadas aos aspectos de controle de projeto. [10].
A gestão de riscos nunca termina (pelo menos em teoria!). A filosofia do gerenciamento de riscos é que não se deve tratar de um conjunto de regras duras e rápidas. O gerenciamento de risco e a estratégia de migração de risco são sobre a compreensão da intenção do gerenciamento de risco e a abordagem lógica e sistemática do processo. Em outras palavras, não basta seguir as regras...pense!
Considering the complexity of medical device design, focused risk management practices help ensure usability, safety, and conformidade regulamentar. It is a process of identifying, controlling, and preventing the failure that may cause hazards to users. It also mandates identifying associated risks.
A Figura 3 mostra todas as etapas envolvidas no processo de gerenciamento de risco. O processo começa com a identificação dos perigos e depois o risco associado é medido com base nas conseqüências dos perigos e sua possibilidade de risco.
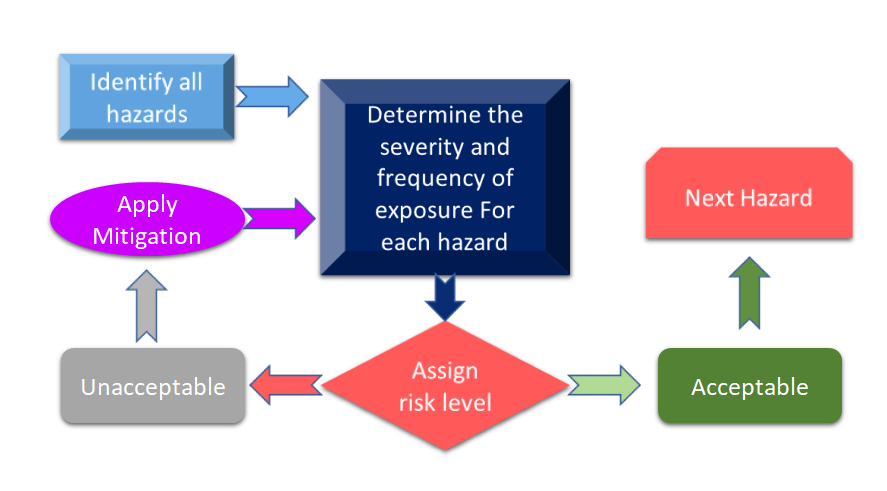
Figura 3: Processo de gerenciamento de risco para Dispositivo Médico (Adaptado de [11]).
Caso o risco identificado durante o processo de gerenciamento de risco para dispositivos médicos esteja acima dos critérios definidos, então você precisará ter uma mitigação de risco. O nível de risco depende de vários parâmetros, incluindo mas não limitado ao seu dispositivo, tecnologias e, em alguns casos, à forma como sua empresa está lidando com o processo de mitigação de risco. É sempre aconselhável conduzir uma análise de risco para seu dispositivo para ver quais padrões podem ser aplicados ao seu dispositivo. Na recente revisão da ISO 14971: A Norma Internacional para Gerenciamento de Risco de Dispositivos Médicos, a análise de risco e a Análise Preliminar de Risco (PHA) são identificadas como exigências principais para seu dispositivo médico [12]. De forma simplificada, o PHA destina-se a fornecer a estrutura inicial para avaliações e gerenciamento de risco e o PHA abrange tanto a análise de risco quanto a avaliação de risco. De acordo com a definição, PHA compreende uma lista de perigos, danos, quaisquer situações perigosas, formulada a partir dos materiais de construção (MoC) de seus dispositivos, componentes ou matéria-prima usada em seu dispositivo, e interfaces homem-dispositivo ou manual, ambiente de uso, princípio de operação e outros fatores relevantes. [13].
Conclusão
No final das contas, para cada inicialização de dispositivo médico ou uma organização bem estabelecida, é importante lembrar que a leitura do regulamento não ganha nada além da compreensão da filosofia que compra muito!
Conclusão: quando se trata de análise de risco e planejamento:
- Deve ser utilizado cedo e durante todo o processo de projeto e desenvolvimento,
- Muitas vezes gerou novas informações para o feedback no processo de projeto e desenvolvimento (à dispositivos atuais/futuros),
- Nenhum planejamento pode eliminar todos os perigos e riscos... mas você pode mitigar muitos deles! (Seguindo as filosofias de controle de projeto aqui descritas mitigam automaticamente os riscos!)
A rota de cada dispositivo médico para o mercado é complexa devido aos vários fatores a serem levados em consideração, tais como padrões de uso, material, experiência do usuário, regulamentos e muito mais.
Precisa de ajuda com medical device design? Browse experienced especialistas da indústria medtech em Kolabtree ou poste seu projeto gratuitamente para receber propostas.
REFERÊNCIAS E RECURSOS
- https://www.welldoc.com/health-plans/
- https://ec.europa.eu/docsroom/documents/10337/attachments/1/translations
- FDA, 2005, Total Product Lifecycle, FDA-CDRH Apresentação pelo Diretor do CDRH Dr. David Feigal, http://www.fda.gov/cdrh/strategic/presentations/ tplc.html
- Pietzsch, Jan & Shluzas, Lauren & Paté-Cornell, Marie-Elisabeth & Yock, Paul & Linehan, John. (2009). Stage-Gate Process for the Development of Medical Devices. Journal of Medical Devices. 3(2).
- Estratégias Regulatórias para a Terceira Edição da IEC 60601-1 Recuperada em 9 de setembro de 2020.
- https://www.meddeviceonline.com/doc/an-introduction-to-international-medical-device-standards-0001
- https://www.fda.gov/files/drugs/published/Design-Controls—Devices.pdf
- Feigal DW. Apêndice D. Impacto do Marco Regulatório no Desenvolvimento e Inovação de Dispositivos Médicos. Comitê do Instituto de Medicina (EUA) sobre o Saúde Pública Effectiveness of the FDA 510(k) Clearance Process; Wizemann T, editor. Public Health Effectiveness of the FDA 510(k) Clearance Process: Balancing Patient Safety and Innovation: Workshop Report. Washington (DC): National Academies Press (US); 2010. Appendix D, Impact of the Regulatory Framework on Medical Device Development and Innovation. Available from: https://www.ncbi.nlm.nih.gov/books/NBK209794/.
- 1997, FDA CDRH 1997, Orientação de Controle de Projeto para Fabricantes de Dispositivos Médicos
- https://starfishmedical.com/blog/iso-134852016-section-7/?doing_wp_cron=1599995964.4528369903564453125000
- Teixeira, M. B., e Bradley, R., 2003, Design Controls for the Medical Device Industry, Marcel Dekker, New York.
- ISO 14971:2019 - Dispositivos médicos - Aplicação da gestão de riscos em dispositivos médicos
- ISO/TR 24971:2020 - Dispositivos médicos - Orientação sobre a aplicação da ISO 14971
Todos os artigos desta série:
Desenvolvimento e Projeto de Dispositivos Médicos: Um Guia Definitivo
Desenvolvimento de Dispositivos Médicos: 3 Dicas para o Sucesso
Projeto de dispositivos médicos: O Essencial, Guia Passo a Passo
Comercialização de Produtos médicos: 9 Passos do Esboço ao Lançamento
Como superar os desafios da comercialização de dispositivos médicos
Lançamento de Dispositivos Médicos: Passos chave para trazer seu produto ao mercado
Vigilância pós-mercado de produtos médicos: Um Guia Abrangente
Kolabtree helps businesses worldwide hire freelance scientists and industry experts on demand. Our freelancers have helped companies publish research papers, develop products, analyze data, and more. It only takes a minute to tell us what you need done and get quotes from experts for free.

Unlock Corporate Benefits
• Secure Payment Assistance
• Onboarding Support
• Dedicated Account Manager
Sign up with your professional email to avail special advances offered against purchase orders, seamless multi-channel payments, and extended support for agreements.


