



An exhaustive guide to conformidade regulamentar for IVD manufacturers, written by Sundeep Agarwal, experiente Consultor do IVDR.
O que é o IVDR?
The European Commission’s (EC) In Vitro Diagnostic Regulation (EU IVDR 2017/746) is a ‘legislative framework’ and a way forward towards global IVD safety, which assures that only reliable and effective IVDs are in the market. The European Commission is trying its best to make the saúde system safer and error free in terms of diagnosis or outcomes.
Os dispositivos médicos de diagnóstico in vitro (IVDD), 98/79/CE foi uma diretiva enquanto IVDR é uma legislação (regulamento) aplicável a todos os Operadores Econômicos (OE), ou seja, fabricantes, importadores, usuários, organismos notificados e autoridades nacionais no Espaço Econômico Europeu (EEE) e aqueles que não são fabricantes e fornecedores da UE que colocam ou planejam distribuir IVD no mercado europeu.
O IVDR consiste em 113 artigos (10 capítulos) e quinze anexos em comparação com 24 artigos, dez anexos do IVDD. Sem dúvida, o IVDR é uma peça de regulamentação longa e consideravelmente rigorosa, mas a parte boa é que, com as mudanças e exigências regulamentares, ele é mais transparente.
Ela enfatiza a abordagem baseada no ciclo de vida. Será aplicada a partir de 26 de maio de 2022 e espera-se que os operadores econômicos (incluindo fabricantes não comunitários) se preparem proativamente para o planejamento e implementação do mesmo. Cada parte interessada no processo será agora igualmente responsável pelo mercado de diagnóstico in-vitro do Espaço Econômico Europeu (EEE).
- A primeira e principal coisa que uma organização deve fazer é organizar um programa de treinamento (on-line ou no local, conforme o caso) no IVDR da UE para que todos na organização estejam cientes das mudanças necessárias.
- Uma comunicação oficial deve ser seguida por, a todos os fornecedores, sub-contratados ou prestadores de serviços sobre o processo e suas obrigações.
- Realizar uma avaliação de lacunas para verificar a disponibilidade de seus recursos, uma equipe competente para atualizar a documentação técnica exigida pelo IVDR da UE. Ter a certificação ISO 13485: 2016 seria uma vantagem adicional para estabelecer a conformidade.
- É aconselhável (se necessário) contratar um especialista no assunto ou um consultor externo desde o estágio inicial da transição, pois "um ponto no tempo economiza nove".
- Este blog fornecerá um esboço detalhado e dicas práticas para atender às expectativas dos organismos notificados e das autoridades competentes, conforme descrito nos vários artigos e anexos do IVDR 2017/746 da UE.
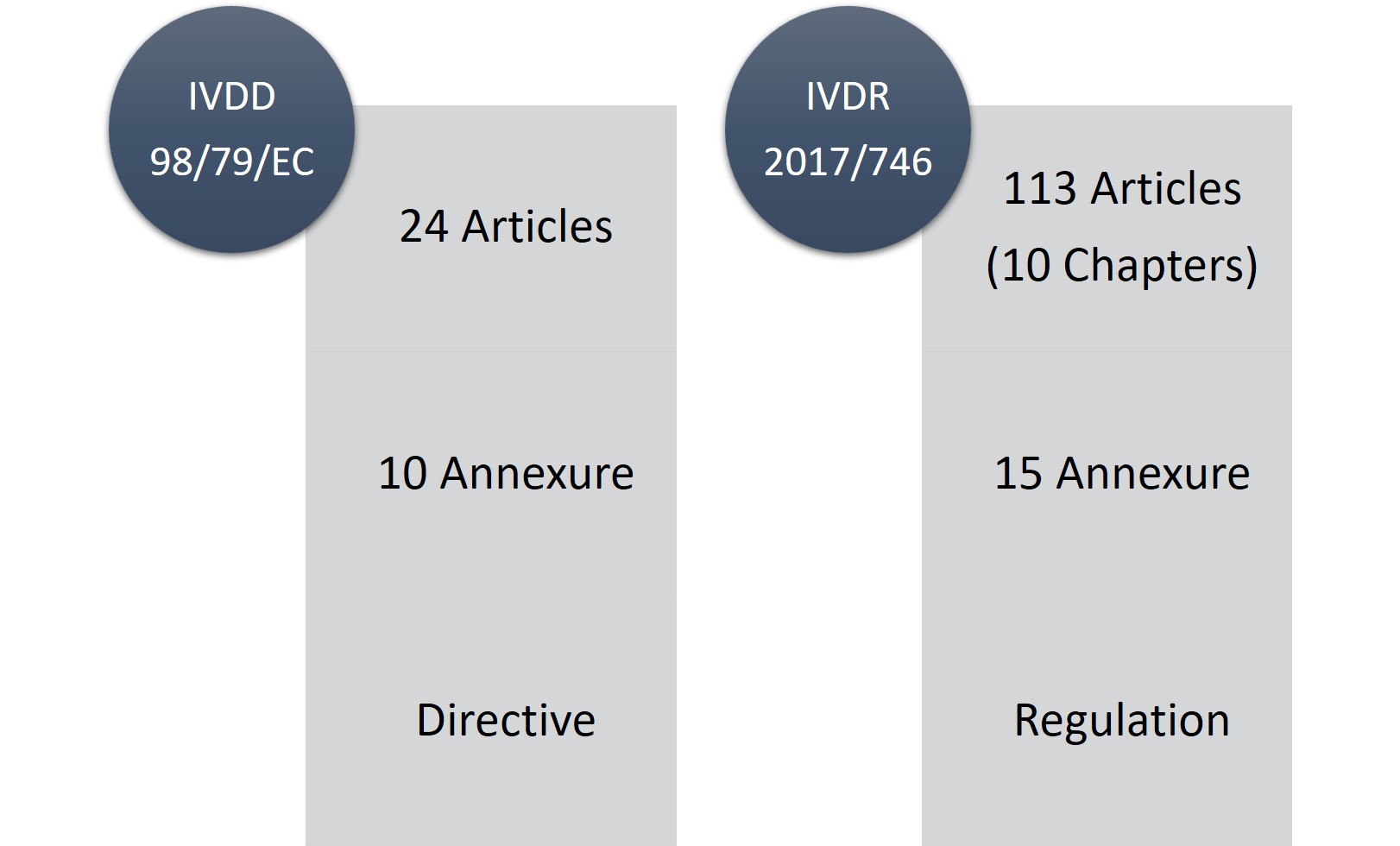
Figura1: IVDD vs. IVDR
1. Preparação para a conformidade IVDR e mudanças comerciais
A principal decisão comercial de uma organização seria concluir se ela quiser continuar a colocar seu DIV no Espaço Econômico Europeu (EEE). Se a resposta for "sim", então deve-se obter estimativas (custo), cronogramas, escopo da auditoria, código de produto, etc. de uma NB o mais cedo possível. Uma mudança de uma diretiva para um regulamento exige uma conformidade obrigatória e uma documentação técnica robusta para estabelecer segurança e eficácia e para obter a Certificação CE. O IVDR se baseia muito mais nas evidências clínicas ou seja, validade científica, desempenho analítico e desempenho clínico para estabelecer a segurança e a eficácia.
O envolvimento de um organismo notificado (NB) no processo de certificação CE será uma característica proeminente do regulamento. Isto também indica um investimento adicional para o operador econômico que pode aumentar indiretamente o custo do produto.
Uma nomeação de um "Pessoa responsável pela conformidade regulamentar (PRRC)". in accordance with Article 15 of EU IVDR 2017/746 is now mandatory; who shall assure the conformity of QMS, declaration of conformity, technical documentation, post market surveillance and reporting of adverse events are in compliance to EU IVDR.Manufacturers should ensure that the entire transition (including new certification application) is completed before the expiry of their existing IVDD Certificate or Self-certified Declaration of conformity. Certificates issued by notified bodies in accordance with IVDD 98/79/EC from 25 May 2017 shall become invalid after 27 May 2024. Be aware of the new timeline for application as per the EC official press release [1] dtd.20th Dezembro de 2022.
2. Compreensão clara da classificação
Reconsiderar a nova regra de classificação sob o Anexo VIII do IVDR e verificar se ela afetou sua classificação anterior.
A realização de uma classificação correta é essencial antes de se preparar para o processo de certificação CE. A menos que sejamos capazes de fazê-lo, o caminho da conformidade será pouco claro e atrasará ou invalidará nossos esforços para cumprir com os requisitos do IVDR.
O IVDR é um abordagem baseada no risco para classificar os dispositivos com o aumento do organismo notificado e dos controles das autoridades competentes. O Regulamento identifica quatro classes de risco: Classe A (menor risco), Classe B, Classe C, e Classe D (maior risco) enquanto o Anexo VIII define sete regras de classificação para classificar corretamente os produtos. Uma característica única do IVDR é que, o Software também é classificado sob a regra de implementação 1.4 do Anexo VIII, que diz "O Software, que dirige um dispositivo ou influencia o uso de um dispositivo, deve se enquadrar na mesma classe que o dispositivo. Se o software for independente de qualquer outro dispositivo, ele deverá ser classificado por direito próprio.[2]”. Isto indica o escopo do Software a ser regulamentado pelo IVDR. E o fabricante também tem que realizar a verificação e validação do software (Anexo II, 6.4) de acordo.
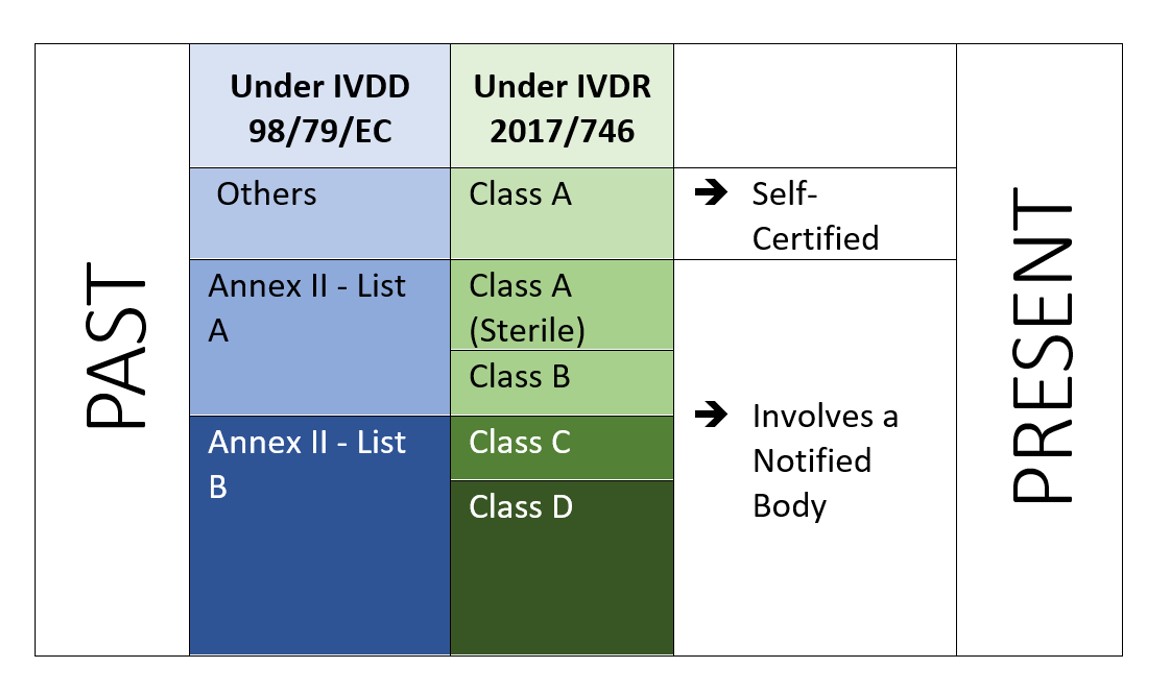
Figura 2: Classificação baseada em risco sob IVDR 2107/746
3. Envolvimento do organismo notificado
O papel de um organismo notificado (NB) seria um dos elementos centrais e, portanto, um número maior de fabricantes exigiria agora ser auditado e certificado por um organismo notificado em relação ao método tradicional de "auto-certificação".Os operadores econômicos devem decidir cuidadosamente a rota de avaliação de conformidade (Anexo IX, X, XI do IVDR da UE[3]).
O IVDR não só exige investimento adicional, mas tem que assegurar que sua documentação técnica e seu sistema de gestão de qualidade atendam às novas exigências do IVDR. Sob o IVDD, a maioria dos IVDs são autocertificados (92%), e não requerem o envolvimento de um Organismo Notificado (exceto 8% do total de IVDs colocados no mercado[4]). Enquanto sob o novo IVDR, o cenário não é o mesmo.
De acordo com um estudo "O impacto das novas regras europeias de classificação IVD sobre o envolvimento do organismo notificado" pelo Instituto Nacional para Saúde Pública e o Meio Ambiente, Bilthoven (Holanda) RIVM Relatório Letter 2018-0082, A. van Drongelen et al., quase 85% de todos os DVDs exigirão o envolvimento do Organismo Notificado, deixando apenas 15% de DVDs elegíveis para autocertificação[5]..
Isto também significa que o fabricante da In Vitro Diagnostics (IVDs) passará agora por uma grande mudança para cumprir com o novo processo de classificação e certificação. Além disso, dependendo do uso pretendido dos dispositivos e da classe de risco, o fabricante precisa identificar um NB designado que possa ser capaz de auditá-los e obter a certificação de seus produtos. O IVD de maior risco (Classe-D) exigiria um laboratório de referência da UE, ou painéis de especialistas para verificar a alegação de desempenho, além do envolvimento de um Organismo Notificado (NB) ou Autoridade Competente (AC). Atualmente, existem apenas seis organismos notificados designados sob o IVDR da UE. Não espere iniciar seu processo de solicitação para evitar atrasos inesperados devido à indisponibilidade de um Organismo Notificado.

Figura 3: Lista de organismos notificados designados pelo IVDR[6]
4. Estabelecimento de um sistema de gestão de qualidade (QMS)
Espera-se que os fabricantes de IVD estabeleçam um sistema de gestão de qualidade (QMS) robusto e confiável dentro de suas instalações. É uma obrigação geral de um fabricante de acordo com o artigo 10 do IVDR. O sistema de gestão de qualidade é um requisito essencial entre vários outros, sem o qual um fabricante não poderá ser aprovado.
QMS is to ensure that manufacturing, change control, customer complaints, resource management, supplier &sub-contractors’ controls and validation, performance evaluation, quality test, UDI Labelling, Post market surveillance etc. are according to approved QMS and Post Market Surveillance (PMS) plans.
O PRRC tem que assegurar que o fabricante tenha cumprido os requisitos do Artigo 10 para "autocertificação" (emissão de Declaração de conformidade de acordo com o Anexo IV) Classe A IVD quando um Organismo Notificado (NB) não for exigido no processo.
5. Estar preparado para a interrupção na cadeia de abastecimento
Throughout the world, manufacturer depends largely on their supply chain and raw material to produce and deliver IVDs that are safe, accurate, and effective forthe intended use. Hence regulatory and quality concerns are also evolving to a higher level when it comes to the suppliers and sub-contractors’ controls. Manufacturer are therefore expected to proactively communicate the supply chain about their obligations and responsibilities of the suppliers and subcontractors. Legal manufacturer shall demonstrate adequate supplier control and monitoring, assure the supply chain is in compliance to the regulatory aspects of IVDR, reconsider the need for data integrity and quality of supplier data, implement robust supplier risk management and performance monitoring and periodically audit the supplier based on the associated risk to the finished products. Os reguladores e organismos notificados estão enfatizando que os fabricantes legais devem documentar claramente o nível de controle dos fornecedores e demonstrar com evidências que eles têm o potencial de mitigar o risco do produto ou serviço fornecido pelo fornecedor.
6. Garantia de auditoria e prontidão de inspeção
De acordo com o artigo 88 do IVDR, Atividades de Vigilância de Mercado, as autoridades competentes devem realizar tanto inspeções anunciadas (não anunciadas) nas instalações dos operadores econômicos como também dos fornecedores e/ou subcontratados e, quando necessário, nas instalações dos usuários profissionais. Enquanto o fabricante deve incluir informações sobre a identificação de todos os locais, incluindo fornecedores e subcontratados, onde as atividades de fabricação são realizadas na Documentação Técnica de Informações de Projeto e Fabricação. Os órgãos notificados (NB) que realizam auditoria de QMS devem identificar links e alocação de responsabilidades entre os vários locais de fabricação, e seus fornecedores e/ou subcontratados. Essas informações serão consideradas enquanto o NB desejar especificamente auditar qualquer um desses fornecedores ou sub-contratados ou ambos. As instalações dos fornecedores do fabricante, quando consideradas como afetando significativamente a conformidade dos dispositivos acabados, devem ser essencialmente auditadas pela NB (em particular quando o fabricante não puder demonstrar controle suficiente sobre seus fornecedores).
7. Plano para lidar com auditorias sem aviso prévio
Sob monitoramento pós-certificação, a NB deve proceder a auditorias sem aviso prévio no local dos fabricantes e seus subcontratados ou fornecedores realizando testes de produtos e o monitoramento do cumprimento de quaisquer condições que vinculem os fabricantes e associadas às decisões de certificação, tais como atualizações de dados clínicos em intervalos definidos.Além disso, torganismo notificado realizará aleatoriamente, pelo menos uma vez a cada cinco anos, auditorias sem aviso prévio no local do fabricante e, quando apropriado, no local dos fornecedores e/ou subcontratados do fabricante, o que pode ser combinado com a avaliação de vigilância periódica.
8. Reforçar as atividades de vigilância pós-mercado
Os fabricantes são fortemente recomendados a fortalecer suas exigências de vigilância pós-comercialização e desenvolver mecanismos de coordenação entre os estados membros da UE sobre vigilância e vigilância de mercado. Sob avaliação de vigilância aplicável aos dispositivos de classe C e classe D (Anexo IX), o organismo notificado deverá periodicamente, pelo menos uma vez a cada 12 meses, realizar auditorias e avaliações apropriadas. Deverá incluir auditorias nas instalações do fabricante e dos fornecedores e/ou subcontratados, conforme aplicável. O fabricante deverá essencialmente desenvolver um procedimento para registro e notificação de incidentes e Ação Corretiva de Segurança de Campo (FSCA).
9. Identificador de dispositivo único (UDI) & EUDAMED
O fabricante deve estabelecer um sistema de UDI para identificar e facilitar a rastreabilidade dos dispositivos. O "Identificador de Dispositivo" e o "Identificador de Produção" devem constar dos rótulos para melhorar a rastreabilidade no mercado da União Européia. Pode-se consultar uma lista de Entidade emissora acreditada (IE) como GS1, HIBCC, ICCBBA, IFA GmbH para operar um sistema para a atribuição de UDIs. Atualmente as IEs mencionadas são válidas a partir de 27th Junho de 2019, mas será sábio confirmar sua validade enquanto se toma uma decisão final sobre sua implementação.
O Banco de Dados Europeu sobre Dispositivos Médicos (EUDAMED) fornecerá uma visão geral de todos os dispositivos médicos disponíveis na União Européia. Ele consiste de seis módulos relacionados a:
- Registro do ator,
- Identificação única do dispositivo (UDI) e registro do dispositivo,
- Órgãos e certificados notificados,
- Investigações clínicas e estudos de desempenho,
- Vigilância e vigilância pós-mercado, e
- Vigilância do mercado.
Para garantir uma maior transparência através de um EUDAMED abrangente, partes das informações dos Operadores Econômicos serão acessíveis ao público. Enquanto as informações confidenciais serão acessíveis somente aos Operadores Econômicos, Patrocinadores, Notificados e Autoridades Competentes dos estados membros da UE.
10. Requisitos para "dispositivos internos".
Health institution developing ‘in-house devices’ (or ‘laboratory-developed tests’) which are meant to be used by the same health institution shall not be marketed or sold to other legal entity. Such devices may be used for the diagnosis and treatment, especially for rare diseases. The institution is expected to comply with only the requirement of Annex I of IVDR (general safety and performance requirements), and exempted from rest of the regulation until 26 May 2024; provided the health institution meets a number of conditions set out in Article 5(5) of the Regulation and has an appropriate quality management system, which complies to the international standard setting out the quality and competence requirements for medical laboratories (EN ISO 15189) or other national provisions, and is able to justify that target patient group’s specific needs cannot appropriately be met by an equivalent device available on the market.
Referências
[1] Comunicado de imprensa oficial da CE dtd. 20th Dez. 2021, Implantação progressiva do Regulamento de Dispositivos Médicos In Vitro Diagnostic. Pode ser acessado em https://ec.europa.eu/commission/presscorner/detail/en/IP_21_6965 [2]REGULAMENTO (UE) 2017/746 DO PARLAMENTO EUROPEU E DO CONSELHO de 5 de abril de 2017 sobre dispositivos médicos de diagnóstico in vitro e que revoga a Diretiva 98/79/CE e a Decisão 2010/227/UE da Comissão; ANEXO VIII REGRAS DE CLASSIFICAÇÃO, 1. REGRAS DE APLICAÇÃO Ponto 1.4 Página 304 [3]Regulamento (UE) 2017/746 do Parlamento Europeu e do Conselho de 5 de abril de 2017 sobre dispositivos médicos de diagnóstico in vitro e revogação da Diretiva 98/79/CE e da Decisão 2010/227/UE da ComissãoANEXO IX Avaliação da conformidade baseada em um sistema de gestão da qualidade e na avaliação da documentação técnica, Página 306, ANEXO X Avaliação da conformidade baseada em exame de tipo, Página 314, ANEXO XI Avaliação da conformidade baseada na garantia da qualidade da produção, Página 317
[4] Comunicado de imprensa dtd. 14 de outubro de 2021, Bruxelas; Saúde pública: A Comissão propõe uma implantação progressiva do novo Regulamento de Dispositivos Médicos In Vitro Diagnósticos [5] O impacto das novas regras européias de classificação dos DIV sobre o envolvimento do organismo notificado: um estudo sobre os DIV registrados na Holanda; van Drongelen A, de Bruijn A, Pennings J, van der Maaden T 32 p em inglês 2018, RIVM letter report 2018-0082 [6] A lista acima é baseada nos dados acessados pela dtd. 5 de março de 2021, para as últimas atualizações na lista, você pode acessar o site oficial do CE em https://ec.europa.eu/growth/tools-databases/nando/index.cfm?fuseaction=directive.notifiedbody&dir_id=35Kolabtree helps businesses worldwide hire freelance scientists and industry experts on demand. Our freelancers have helped companies publish research papers, develop products, analyze data, and more. It only takes a minute to tell us what you need done and get quotes from experts for free.

Unlock Corporate Benefits
• Secure Payment Assistance
• Onboarding Support
• Dedicated Account Manager
Sign up with your professional email to avail special advances offered against purchase orders, seamless multi-channel payments, and extended support for agreements.


