



Dr. Javier Quilez Oliete, um experiente freelance bioinformatics consultant on Kolabtree, provides a comprehensive guide to DNA sequencing análise de dados, including tools and software used to read data.
Introdução
O ácido desoxirribonucleico (DNA) é a molécula que transporta a maior parte da informação genética de um organismo. (Em alguns tipos de vírus, a informação genética é transportada pelo ácido ribonucleico (RNA)). Os nucleotídeos (convencionalmente representados pelas letras A, C, G ou T) são as unidades básicas das moléculas de DNA. Conceitualmente, Seqüenciamento de DNA é o processo de leitura dos nucleotídeos que compõem uma molécula de DNA (por exemplo, "GCAAACCAAT" é uma cadeia de 10 nucleotídeos de DNA). As tecnologias atuais de seqüenciamento produzem milhões de leituras de DNA deste tipo. em um tempo razoável e a um custo relativamente baixo. Como referência, o custo de seqüenciar um genoma humano - um genoma é o conjunto completo de moléculas de DNA em um organismo - diminuiu o $100 barreira e isso pode ser feito em questão de dias. Isto contrasta com a primeira iniciativa de seqüenciar o genoma humanoO projeto, que foi concluído em uma década e teve um custo de cerca de $2,7 bilhões.
This capability to sequence DNA at high throughput and low cost has enabled the development of a growing number of sequencing-based methods and applications. For example, sequencing entire genomes or their protein-coding regions (two approaches known respectively as whole genome and exome sequencing) in disease and healthy individuals can hint to disease-causing DNA alterations. Also, the sequencing of the RNA that is transcribed from DNA—a technique known as RNA-sequencing—is used to quantify gene activity and how this changes in different conditions (e.g. untreated versus treatment). On the other side, chromosome conformation capture sequencing methods detect interactions between nearby DNA molecules and thus help to determine the spatial distribution of chromosomes within the cell.
Comum a estas e outras aplicações de seqüenciamento de DNA é a geração de conjuntos de dados na ordem dos gigabytes e compreendendo milhões de seqüências de leitura. Portanto, fazer sentido para os experimentos de seqüenciamento de alta produção (HTS) requer capacidades substanciais de análise de dados. Felizmente, existem ferramentas computacionais e estatísticas dedicadas e fluxos de trabalho de análise relativamente padrão para a maioria dos tipos de dados HTS. Embora algumas das etapas (iniciais) de análise sejam comuns à maioria dos tipos de dados de seqüenciamento, mais análise downstream dependerá do tipo de dados e/ou do objetivo final da análise. A seguir, forneço uma cartilha sobre as etapas fundamentais na análise dos dados HTS e me refiro a ferramentas populares.
Algumas das seções abaixo estão focadas na análise dos dados gerados a partir de tecnologias de sequenciamento de leitura curta (em sua maioria Illumina), já que estes dominaram historicamente o mercado HTS. Entretanto, tecnologias mais novas que geram leituras mais longas (por exemplo Oxford Nanopore Technologies, PacBio) estão ganhando terreno rapidamente. Como o sequenciamento de leitura longa tem algumas particularidades (por exemplo, taxas de erro mais altas), ferramentas específicas estão sendo desenvolvidas para a análise deste tipo de dados.
Controle de qualidade (QC) de leituras brutas
O analista ansioso iniciará a análise a partir dos arquivos FASTQ; o Formato FASTQ tem sido por muito tempo o padrão para armazenar dados de seqüenciamento de leitura curta. Em essência, os arquivos FASTQ contêm a seqüência de nucleotídeos e a seqüência por base chamando qualidade para milhões de leituras. Embora o tamanho do arquivo dependa do número real de leituras, os arquivos FASTQ são tipicamente grandes (na ordem de megabytes e gigabytes) e comprimidos. Note que a maioria das ferramentas que usam arquivos FASTQ como entrada podem manuseá-los em formato comprimido, portanto, para economizar espaço em disco, é recomendável não descomprimi-los. Como uma convenção, aqui vou equiparar um arquivo FASTQ a uma amostra sequencial.
FastQC é provavelmente a ferramenta mais popular para realizar o CQ das leituras em bruto. Ela pode ser executada através de uma interface visual ou programática. Enquanto a primeira opção pode ser mais conveniente para usuários que não se sentem confortáveis com o ambiente de linha de comando, a segunda oferece escalabilidade e reprodutibilidade incomparáveis (pense no quão tedioso e propenso a erros pode ser executar manualmente a ferramenta para dezenas de arquivos). De qualquer forma, a principal saída do FastQC é um Arquivo HTML reporting key summary estatísticas about the overall quality of the raw sequencing reads from a given sample. Inspecting tens of FastQC reports one by one is tedious and it complicates the comparison across samples. Therefore, you may want to use MultiQCque agrega os relatórios HTML do FastQC (assim como de outras ferramentas utilizadas a jusante, por exemplo, corte do adaptador, alinhamento) em um único relatório.
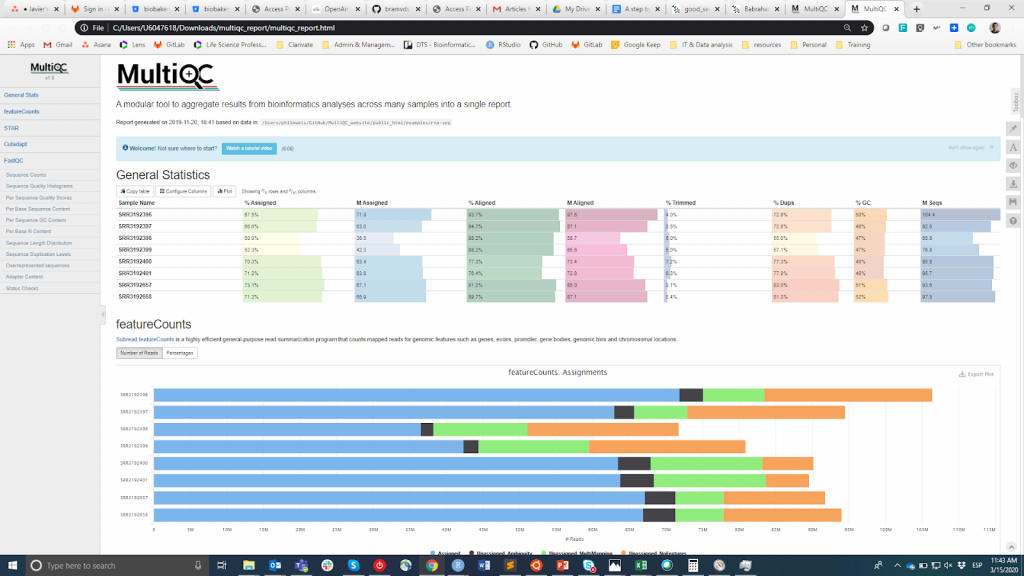
MultiQC
As informações de CQ destinam-se a permitir ao usuário julgar se as amostras têm boa qualidade e podem, portanto, ser utilizadas para as etapas subseqüentes ou se precisam ser descartadas. Infelizmente, não há um limiar de consenso baseado na métrica FastQC para classificar as amostras como de boa ou má qualidade. A abordagem que eu utilizo é a seguinte. Espero que todas as amostras que passaram pelo mesmo procedimento (por exemplo, extração de DNA, preparação da biblioteca) tenham estatísticas de qualidade semelhantes e uma maioria de bandeiras de "aprovação". Se algumas amostras tiverem qualidade inferior à média, eu ainda as usarei na análise a jusante tendo isto em mente. Por outro lado, se todas as amostras da experiência receberem sistematicamente bandeiras de "aviso" ou "falha" em múltiplas métricas (ver este exemplo), suspeito que algo deu errado na experiência (por exemplo, má qualidade do DNA, preparação da biblioteca, etc.) e recomendo que seja repetido.
Leia o recorte
O CQ de leituras em bruto ajuda a identificar amostras problemáticas, mas não melhora a qualidade real das leituras. Para fazer isso, precisamos aparar as leituras para remover seqüências técnicas e fins de baixa qualidade.
Seqüências técnicas são sobras do procedimento experimental (por exemplo, adaptadores de seqüenciamento). Se tais seqüências forem adjacentes à seqüência verdadeira da leitura, o alinhamento (ver abaixo) pode mapear leituras para a posição errada no genoma ou diminuir a confiança em um determinado alinhamento. Além das seqüências técnicas, podemos também querer remover seqüências de origem biológica se estas estiverem altamente presentes entre as leituras. Por exemplo, procedimentos subótimos de preparação do DNA podem deixar uma alta proporção de RNA ribossômico convertido em DNA (rRNA) na amostra. A menos que este tipo de ácido nucleico seja o alvo do experimento de seqüenciamento, manter leituras derivadas do rRNA apenas aumentará a carga computacional das etapas a jusante e pode confundir os resultados. Note que, se os níveis de seqüências técnicas, rRNA ou outro contaminante forem muito altos, o que provavelmente já terá sido destacado pelo CQ, você pode querer descartar toda a amostra sequenciada.
No sequenciamento de leitura curta, a seqüência de DNA é determinada um nucleotídeo de cada vez (tecnicamente, um nucleotídeo a cada ciclo de sequenciamento). Em outras palavras, o número de ciclos de seqüenciamento determina a duração da leitura. Uma questão conhecida dos métodos de seqüenciamento HTS é a decadência da precisão com que os nucleotídeos são determinados à medida que os ciclos de seqüenciamento se acumulam. Isto se reflete em uma diminuição geral da qualidade de chamada por base, especialmente no final da leitura. Como acontece com as seqüências técnicas, tentar alinhar leituras que contêm pontas de baixa qualidade pode levar a uma má colocação ou má qualidade de mapeamento.
Para remover seqüências técnicas/contaminantes e fins de baixa qualidade, leia ferramentas de corte como Trimmomatic e Cutadapt existem e são amplamente utilizadas. Em essência, tais ferramentas removerão seqüências técnicas (disponíveis internamente e/ou fornecidas pelo usuário) e leituras de acabamento baseadas na qualidade enquanto maximizam o comprimento de leitura. As leituras que são deixadas muito curtas após o corte são descartadas (leituras excessivamente curtas, por exemplo, <36 nucleotídeos, complicam a etapa de alinhamento, já que estes provavelmente mapearão para múltiplos locais no genoma). Você pode querer olhar a porcentagem de leituras que sobrevivem ao corte, pois uma alta taxa de leituras descartadas é provavelmente um sinal de dados de má qualidade.
Finalmente, eu normalmente volto a executar FastQC nas leituras aparadas para verificar se este passo foi eficaz e melhorou sistematicamente as métricas de QC.
Alinhamento
Com exceções (por exemplo de novo assembléia), o alinhamento (também referido como mapeamento) é normalmente o próximo passo para a maioria dos tipos de dados e aplicações HTS. O alinhamento de leitura consiste em determinar a posição no genoma do qual deriva a seqüência de leitura (tipicamente expressa como cromossomo: start-end). Portanto, nesta etapa exigimos o uso de uma seqüência de referência para alinhar/mapear as leituras.
A escolha da seqüência de referência será determinada por múltiplos fatores. Para um, a espécie da qual o DNA sequenciado é derivado. Embora o número de espécies com uma seqüência de referência de alta qualidade disponível esteja aumentando, este pode ainda não ser o caso para alguns organismos menos estudados. Nesses casos, você pode querer alinhar leituras com uma espécie evolutivamente próxima para a qual um genoma de referência está disponível. Por exemplo, como não há uma seqüência de referência para o genoma do coiote, podemos usar a do cão estreitamente relacionado para o alinhamento de leitura. Da mesma forma, podemos ainda querer alinhar nossas leituras a uma espécie intimamente relacionada para a qual existe uma seqüência de referência de maior qualidade. Por exemplo, enquanto o genoma do gibão tem sido publicadoA organização do genoma é dividida em milhares de fragmentos que não recapitulam completamente a organização daquele genoma em dezenas de cromossomos; nesse caso, realizar o alinhamento usando a seqüência de referência humana pode ser benéfico.
Outro fator a ser considerado é a versão da montagem da seqüência de referência, uma vez que novas versões são lançadas à medida que a seqüência é atualizada e melhorada. É importante notar que as coordenadas de um determinado alinhamento podem variar entre as versões. Por exemplo, várias versões do genoma humano podem ser encontradas no Navegador de Genoma da UCSC. Em qualquer espécie, eu sou fortemente a favor da migração para a mais nova versão de montagem, uma vez que esta seja totalmente liberada. Isto pode causar alguns transtornos durante a transição, pois os resultados já existentes serão relativos às versões mais antigas, mas compensa a longo prazo.
Além disso, o tipo de dados de seqüenciamento também importa. As leituras geradas a partir dos protocolos DNA-seq, ChIP-seq ou Hi-C serão alinhadas à seqüência de referência do genoma. Por outro lado, como o RNA transcrito do DNA é processado posteriormente em mRNA (ou seja, introns removidos), muitas leituras de RNA-seq falharão no alinhamento com uma seqüência de referência do genoma. Em vez disso, precisamos alinhá-los para transcriptoma seqüências de referência ou usar alinhadores sensíveis à divisão (veja abaixo) ao usar a seqüência de genoma como referência. Relacionado a isto está a escolha da fonte para a anotação da seqüência de referência, ou seja, o banco de dados com as coordenadas dos genes, transcrições, centrômeros, etc. Eu normalmente uso o Anotação GENCODE pois combina anotações genéticas abrangentes e seqüências de transcrição.
Uma longa lista de ferramentas de alinhamento de sequências de leitura curta foi desenvolvida (veja a seção de alinhamento de sequências de leitura curta aqui). Reviewing them is beyond the scope of this article (details about the algorithms behind these tools can be found aqui). Em minha experiência, entre as mais populares estão Bowtie2, BWA, HISAT2, Minimap2, STAR e TopHat. Minha recomendação é que você escolha seu alinhador com base em fatores chave como o tipo de dados HTS e aplicação, assim como aceitação pela comunidade, qualidade da documentação e número de usuários. Por exemplo, são necessários alinhadores como STAR ou Bowtie2 que estejam conscientes das junções exon-exon ao mapear o RNA-seq para o genoma.
Comum à maioria dos mapeadores é a necessidade de indexar a seqüência utilizada como referência antes que o alinhamento real ocorra. Esta etapa pode ser demorada, mas só precisa ser feita uma vez para cada seqüência de referência. A maioria dos mapeadores irá armazenar alinhamentos em arquivos SAM/BAM, que seguem a Formato SAM/BAM (Arquivos BAM são versões binárias de arquivos SAM). O alinhamento está entre as etapas mais demoradas e computacionais na análise dos dados de seqüenciamento e os arquivos SAM/BAM são pesados (na ordem de gigabytes). Portanto, é importante garantir que você tenha os recursos necessários (veja a seção final abaixo) para executar o alinhamento em um tempo razoável e armazenar os resultados. Da mesma forma, devido ao tamanho e formato binário dos arquivos BAM, evite abri-los com editores de texto; em vez disso, use comandos Unix ou ferramentas dedicadas como SAMtools.
A partir dos alinhamentos
Eu diria que não há um claro passo comum após o alinhamento, pois neste ponto é onde cada tipo de dado HTS e aplicação pode diferir.
Uma análise downstream comum para dados de DNA-seq é a chamada variante, ou seja, a identificação de posições no genoma que variam em relação à referência do genoma e entre indivíduos. Uma estrutura de análise popular para esta aplicação é GATK para polimorfismo de nucleotídeos simples (SNP) ou pequenas inserções/deleções (indels) (Figura 2). As variantes que compreendem pedaços maiores de DNA (também referidas como variantes estruturais) requerem métodos de chamada dedicados (ver este artigo para uma comparação abrangente). Como com os alinhadores, aconselho selecionar a ferramenta correta considerando fatores-chave como o tipo de variantes (SNP, indel ou variantes estruturais), aceitação pela comunidade, qualidade da documentação e número de usuários.
Provavelmente a aplicação mais freqüente do RNA-seq é a quantificação da expressão gênica. Historicamente, as leituras precisavam ser alinhadas à seqüência de referência e então o número de leituras alinhadas a um determinado gene ou transcrição era usado como um proxy para quantificar seus níveis de expressão. Esta abordagem de alinhamento+quantificação é realizada por ferramentas como Botões de punho, RSEM ou característicasCounts. Entretanto, a abordagem scuh tem sido cada vez mais superada por novos métodos implementados em software, como Kallisto e Salmão. Conceptualmente, com tais ferramentas, a seqüência completa de uma leitura não precisa ser alinhada com a seqüência de referência. Em vez disso, precisamos apenas alinhar nucleotídeos suficientes para ter certeza de que uma leitura se originou de uma determinada transcrição. Dito de forma simples, a abordagem alinhamento+quantificação é reduzida a uma única etapa. Esta abordagem é conhecida como pseudo-mapeamento e aumenta muito a velocidade da quantificação da expressão gênica. Por outro lado, tenha em mente que o pseudo-mapeamento não será adequado para aplicações onde o alinhamento completo é necessário (por exemplo, chamada de variante a partir de dados do RNA-seq).
Outro exemplo das diferenças nas etapas de análise downstream e as ferramentas necessárias para a aplicação baseada em seqüenciamento é o ChIP-seq. As leituras geradas com tal técnica serão usadas para a chamada de pico, que consiste em detectar regiões no genoma com um excesso significativo de leituras que indicam onde a proteína alvo está ligada. Existem vários picos de chamada e esta publicação os pesquisa. Como exemplo final mencionarei os dados Hi-C, nos quais são usados alinhamentos como entrada para ferramentas que determinam as matrizes de interação e, a partir delas, as características 3D do genoma. Comentando todos os ensaios baseados em seqüenciamento além do escopo deste artigo (para uma lista relativamente completa, veja este artigo).
Antes de começar...
A parte restante deste artigo toca em aspectos que podem não ser estritamente considerados como passos na análise dos dados HTS e que são em grande parte ignorados. Em contraste, eu argumento que é capital que você pense sobre as questões colocadas em Tabela 1 antes de começar a analisar os dados HTS (ou qualquer tipo de dado de fato), e eu escrevi sobre estes tópicos aqui e aqui.
Tabela 1
| Pense sobre isso | Ação proposta |
| Você tem todas as informações de sua amostra necessárias para a análise? | Coletar sistematicamente os metadados dos experimentos |
| Você será capaz de identificar inequivocamente sua amostra? | Estabelecer um sistema para atribuir a cada amostra um identificador único |
| Onde estarão os dados e os resultados? | Organização estruturada e hierárquica dos dados |
| Você será capaz de processar várias amostras sem problemas? | Escalabilidade, paralelização, configuração automática e modularidade do código |
| Você ou qualquer outra pessoa será capaz de reproduzir os resultados? | Documente seu código e procedimentos! |
Como mencionado acima, os dados brutos do HTS e alguns dos arquivos gerados durante suas análises estão na ordem de gigabytes, portanto não é excepcional que um projeto que inclua dezenas de amostras exija terabytes de armazenamento. Além disso, algumas etapas na análise dos dados HTS são computacionalmente intensivas (por exemplo, alinhamento). Entretanto, a infra-estrutura de armazenamento e computação necessária para a análise de dados HTS é uma consideração importante e muitas vezes é negligenciada ou não discutida. Como exemplo, como parte de uma análise recente, revisamos dezenas de artigos publicados que realizam análise de associação fenomérica (PheWAS). O PheWAS moderno analisa 100-1.000s tanto de variantes genéticas quanto de fenótipos, o que resulta em um importante armazenamento de dados e poder computacional. E ainda assim, praticamente nenhum dos artigos que revisamos comentou sobre a infra-estrutura necessária para a análise PheWAS. Não surpreendentemente, minha recomendação é que você planeje com antecedência os requisitos de armazenamento e computação que enfrentará e os compartilhe com a comunidade.
Precisa de ajuda na análise dos dados de seqüenciamento de DNA? Entre em contato com freelance bioinformatics specialist e especialistas em genômica em Kolabtree.
Kolabtree helps businesses worldwide hire freelance scientists and industry experts on demand. Our freelancers have helped companies publish research papers, develop products, analyze data, and more. It only takes a minute to tell us what you need done and get quotes from experts for free.

Unlock Corporate Benefits
• Secure Payment Assistance
• Onboarding Support
• Dedicated Account Manager
Sign up with your professional email to avail special advances offered against purchase orders, seamless multi-channel payments, and extended support for agreements.


