



Shreya Chenni, redator regulador freelance for medical devices, provides a 10-minute guide to FDA design controls for your dispositivo médico.
Uma das principais causas de recalls de dispositivos médicos é a falta de controle do projeto, conforme identificado pela FDA [3,3a]. Os controles de pré-produção foram então acrescentados às normas GMP do dispositivo. Os controles de projeto são um conjunto inter-relacionado de práticas e procedimentos que são incorporados ao processo de projeto e desenvolvimento, ou seja, um sistema de verificações e balanços. Os controles de projeto aumentam a probabilidade de que o projeto transferido para a produção se traduza em um dispositivo que seja apropriado para seu uso pretendido.
Aplicabilidade
Todos os dispositivos Classe II, III e os seguintes Classe I estão sujeitos a controles de projeto:
- Devices automated with computer software
- 868.6810 Cateter, Sucção Traqueobrônquica
- 878.4460 Luva, Cirurgião
- 880.6760 Restrição, Protetor
- 892.5650 Sistema, Aplicador, Radionuclídeo, Manual
- 892.5740 Fonte, Radionuclídeo Teleterapia
- Dispositivos automatizados com software de computador
- Cateteres de sucção traqueobrônquica
- Luvas de cirurgião
- Restrições protetoras
- Sistema, radionuclídeo, aplicador, manual
- Fonte, teleterapia por radionuclídeos
Design controls apply to all D&D activities – for novel or improved devices being developed in the pre-market phase, as well as for changes to existing, marketed devices. Design controls do not apply to pesquisa activities conducted during the proof of concept stage.
Design controls can be applied to any desenvolvimento de produtos process. The following flowcharts are examples of the design controls applied to a traditional Waterfall design process and V-model process for Software (SW).
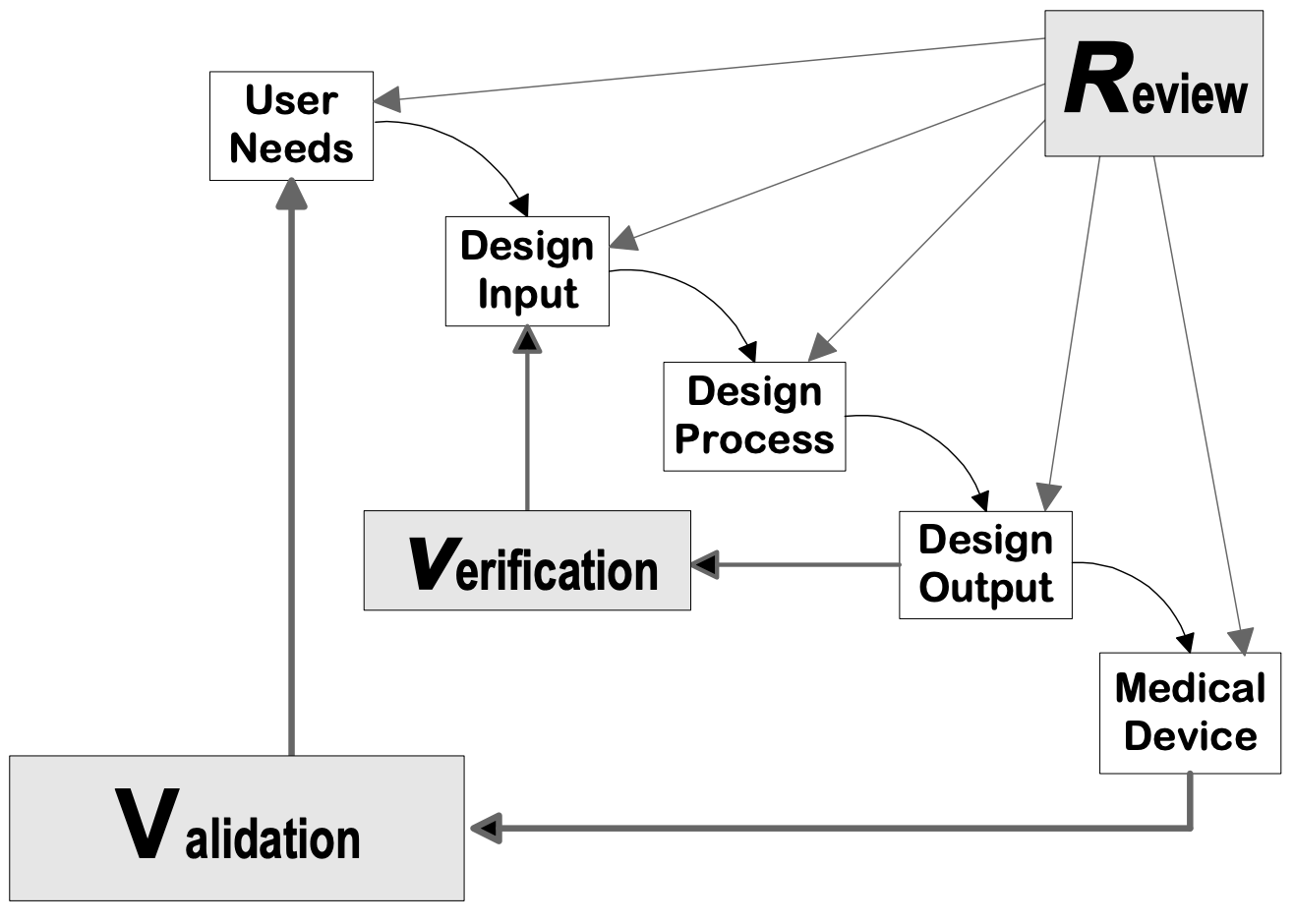
Fonte: Orientação de Controle de Projeto para Fabricantes de Dispositivos Médicos, CDRH-FDA
Fases de projeto do dispositivo
Fase de planejamento D&D
Os planos de D&D devem ser estabelecidos e mantidos. O plano deve descrever ou referenciar as atividades de projeto e desenvolvimento e atribuir responsabilidades pela implementação. No mínimo, a FDA recomenda a inclusão do seguinte no plano:
- Metas e objetivos do programa D&D
- Delimitação das responsabilidades das atividades de projeto
- Identificar as principais tarefas, entregar os produtos e designar a responsabilidade por cada tarefa
- Programação da tarefa principal de acordo com o principal cronograma de desenvolvimento
- Identificar as principais revisões e pontos de decisão
- Identificar os revisores, a equipe de revisão e os procedimentos a serem seguidos pelos revisores
- Controles da documentação do projeto
- Atividades de notificação
O plano deve ser revisto, atualizado e aprovado à medida que o projeto e o desenvolvimento evoluem.
Fase de entrada do projeto
This is the starting point for product design. The medical device is designed and developed to meet the user requirements. Gather the user requirements from various sources such as customer surveys, feedback from the physicians, complaints. These requirements are transferred into design inputs.
- Insumos de projeto: Estes são os requisitos físicos e de desempenho de um dispositivo que são usados para o projeto de um dispositivo. A fase de entrada do projeto consiste na conversão das exigências do usuário em exigências do produto. Os requisitos regulamentares são considerados ao definir as entradas de projeto. Os requisitos de entrada do projeto devem ser abrangentes, sem ambigüidade e verificáveis objetivamente. Os requisitos de entrada podem ser agrupados em 3 categorias:
- Exigências funcionais, que descrevem o que o dispositivo faz. Por exemplo, o que o dispositivo faz: A cadeira de rodas deve seguir em frente quando indicada pelo usuário
- Os requisitos de desempenho, que especificarão quanto e quão bem o dispositivo deve funcionar. Por exemplo: A cadeira de rodas deve mover-se com uma velocidade de 2m/s na direção da frente
- Requisitos de interface, especificar as características do dispositivo que são críticas para a compatibilidade com sistemas externos, tais como interface usuário/paciente. Por exemplo: A cadeira de rodas deve ter botões com símbolos indicativos de direção
- Aqui está um exemplo, que define as necessidades do usuário e as converte para a entrada do projeto:
- Necessidade do usuário
O dispositivo deve ser portátil e com bluetooth habilitado
- Necessidade do usuário
-
- Entrada de projeto
Identificar o FDA aplicável reconhecido ou uma norma internacional a ser cumprida. Tais como:
-
-
- IEEE ANSI C63.27-2017 Norma Nacional Americana para Avaliação da Coexistência Sem Fio
- AAMI TIR 69: Association for the Advancement of Medical Instrumentation – Gerenciamento de risco of Radio-frequency Wireless Coexistence for Medical Devices and Systems (2017)
- IEC 60601-1-2 Edição 3: 2007: Equipamento Elétrico Médico - Parte 1-2: Requisitos Gerais de Segurança - Norma Colateral: Compatibilidade Eletromagnética - Requisitos e Testes
- UL 2054 - Norma para Baterias Domésticas e Comerciais
-
Liste o desempenho e outros insumos específicos. Como, por exemplo:
-
-
- O dispositivo deve ser alimentado por corrente contínua
- O módulo Bluetooth deve ser utilizado
- Peso: aproximadamente 6lbs ou 6lbs+/- 2lbs (As entradas devem ter limites quantitativos para garantir a verificação)
-
Fase de projeto de saída
Os resultados do projeto são o resultado de um esforço de projeto em cada fase do projeto e no final do esforço total de projeto. Exemplos de saídas de projeto são desenhos de engenharia, etiquetagem, instruções de trabalho e outras especificações do produto. Outras saídas de projeto incluem resultados de análise de risco, resultados de atividades de verificação, resultados de testes de biocompatibilidade e código fonte do software.
Os resultados do projeto não devem ser liberados antes da revisão e aprovação pelo pessoal responsável. Também deve ser observado que quaisquer mudanças no dispositivo após a aprovação das saídas/inputs do projeto serão controladas através de revisão e aprovação pelo pessoal envolvido. Uma revisão do projeto é necessária no final desta fase.
Fase de revisão do projeto
Uma revisão do projeto deve ser realizada após a fase de saída do projeto. A revisão do projeto deve seguir os procedimentos estabelecidos e ser documentada no Arquivo de Histórico de Projeto (DHF). Os participantes de cada revisão de projeto devem ter representantes de todos os grupos funcionais.
Recomenda-se a realização de revisões formais no final de marcos importantes do projeto. Geralmente, as revisões de projeto são conduzidas após a fase de saída do projeto, fase de V&V e transferência de projeto phanse. Isto também depende da complexidade do desenvolvimento do dispositivo. A FDA exige pelo menos uma revisão do projeto.
Fase de verificação do projeto
A verificação do projeto é a confirmação pela evidência objetiva de que a saída do projeto corresponde à entrada do projeto. Basicamente, é Design Input = Design Output. As atividades de verificação devem ser realizadas de acordo com os procedimentos estabelecidos. Exemplos de verificação incluem testes EMC e elétricos, inspeção visual, atividades de testes não-clínicos, análise de árvore de falhas do processo ou projeto e análise de modos e efeitos de falhas. A verificação assegura que os requisitos técnicos da especificação do produto sejam atendidos. Todas as atividades de verificação têm de ser documentadas.
Matriz de Rastreabilidade: Este documento consiste em entradas e saídas de projeto listadas em um formato tabular. Para cada entrada, a saída correspondente é referenciada. Este método de verificação é usado quando as entradas e saídas são ambos documentos.
Fase de validação do projeto
A validação do projeto significa estabelecer, através de provas objetivas, que as especificações (requisitos especificados) estão em conformidade com as necessidades do usuário e o(s) uso(s) pretendido(s).
- Validação do processo significa estabelecer, através de evidências objetivas, que um processo produz consistentemente um resultado ou produto que atende a suas especificações pré-determinadas.
- A validação do projeto significa estabelecer, por evidência objetiva, que as especificações do dispositivo estão em conformidade com as necessidades do usuário e o(s) uso(s) previsto(s).
A validação é tipicamente realizado sob condições reais ou simuladas. Examples of validation include ensaios clínicos, clinical evaluation, human factors tests, address packaging and labeling, analysis and inspections. Results of validation activities and/or validation reports should be documented which will be part of the Design History File (DHF).
Fase de transferência do projeto
Após a conclusão da etapa de V&V, ocorre a transferência do projeto. Isto inclui a transferência do projeto do dispositivo para as especificações do produto, garantindo a qualidade do dispositivo. Esta fase é muito crítica porque, uma vez iniciada a produção do dispositivo, ele será submetido a um controle de mudança de projeto e pode resultar em perdas financeiras se forem encontrados problemas. A transferência do projeto deve ocorrer de acordo com o procedimento estabelecido. Além disso, deve-se assegurar que os documentos que incluem especificações de produto sejam revisados e aprovados antes de iniciar a transferência do projeto.
Fase de mudanças de projeto
O controle de mudança de projeto começa com a transferência do projeto e continua durante todo o ciclo de vida. Qualquer mudança no projeto após a transferência do projeto resultará em um Aviso de Mudança de Projeto (ECN) a ser realizado de acordo com um procedimento estabelecido. Deve-se assegurar que com qualquer mudança no projeto, documentos relacionados, tais como relatório de gerenciamento de risco, instruções de uso, relatórios de verificação e validação também devem ser revisados e atualizados.
Arquivo de História do Projeto (DHF)
O DHF é específico para a FDA dos EUA. A ISO 13486:2016 não exige que o fabricante mantenha um DHF.
Para cada projeto é mantido um Arquivo de Histórico de Projeto que inclui todos os resultados de cada fase. Ele inclui as informações mais recentes do produto. Os documentos de projeto e desenvolvimento devem estar facilmente disponíveis e acessíveis conforme e quando necessário. Os contratos de projeto e desenvolvimento devem especificar explicitamente o direito do fabricante a informações de projeto e estabelecer padrões para a forma e conteúdo da documentação de projeto.
Na prática, os controles de projeto proporcionam aos gerentes e projetistas uma melhor visibilidade do processo de projeto. Com maior visibilidade, os gerentes têm o poder de dirigir mais efetivamente o processo de projeto, ou seja, reconhecer problemas mais cedo, fazer correções e ajustar a alocação de recursos. Os projetistas se beneficiam tanto pela melhor compreensão do grau de conformidade de um projeto com as necessidades dos usuários e pacientes, quanto por uma melhor comunicação e coordenação entre todos os participantes do processo.
Precisa de ajuda para entender e implementar os Controles de Projeto da FDA para seu dispositivo médico? Contato experiente consultores em dispositivos médicos em Kolabtree.
Referências:
- FDA, Orientação de Controle de Projeto para Fabricantes de Dispositivos Médicos
- Práticas de regulamentação médica, Uma perspectiva internacional, Val Theisz
- Design Control, apresentação de Joseph Tartal
3a. Registro Federal / Vol. 61, No. 195
Kolabtree helps businesses worldwide hire freelance scientists and industry experts on demand. Our freelancers have helped companies publish research papers, develop products, analyze data, and more. It only takes a minute to tell us what you need done and get quotes from experts for free.

Unlock Corporate Benefits
• Secure Payment Assistance
• Onboarding Support
• Dedicated Account Manager
Sign up with your professional email to avail special advances offered against purchase orders, seamless multi-channel payments, and extended support for agreements.


