



Biólogo Computacional e Kolabtree freelancer Shaurya Jauhari writes about current challenges involved in conducting pathway analysis in bioinformatics and possible solutions to the problem.
In the contrasting era of “Imprecise Medicine” (propelling us to tune towards Precision Medicine) and inundation of biomedical data brought forth by advancements in instrumentation technologies, a lacuna persists that is largely premised over mapping data to information. The clinical experiments engender biomarcadores (technically list of genes or genomic regions more contemporarily) that have to be expounded for their biological implications. The current suite of tools that facilitate such an endeavor is less purposeful as it neglects the ipso factoA maioria das células eucarióticas é constituída por um genoma, interações espaciais do genoma dado seu perfil de acomodação dentro do núcleo de cada célula eucariótica. Este comentário trata de destacar a natureza do problema, lançar luz sobre a organização do genoma, refletir brevemente sobre as ferramentas de mapeamento atuais e conjecturar as possíveis soluções.
O diabo está no detalhe
Uma série de esforços no sentido de aprimorar a resolução dos dados genômicos está escovando debaixo de um esquema crucial de informação. Estamos impulsionando ansiosamente a probabilidade de um genoma $1000, embora nos preocupemos menos com a análise do $100.000. Existe um grande compêndio de repertórios que contém as anotações dos resultados experimentais e os casos sob um estudo biológico típico. Poderia haver definições declarando as implicações biológicas de um gene, ou o caminho do qual esses genes fazem parte, estando associados a uma doença. Mais uma vez, este tipo de armazenamento dinâmico e informativo foi curado manualmente (enquanto isso) e a gestão do conhecimento foi assumida por oleodutos automatizados que empregam computadores e ICT em geral. Estas bases de dados são atualizadas com uma sabedoria científica consensual e têm adotado um punhado de revisões desde o início. O mapeamento dos resultados experimentais para suas implicações biológicas é fortemente subjugado, em grande parte porque a "verdadeira" biologia subjacente é descartada.
Nosso genoma, com cerca de 2 metros de comprimento em média, é acomodado dentro do núcleo de cada célula que reveste nosso corpo. Devido à estrutura diminuta de uma célula e mais do seu núcleo, o genoma é embalado de uma forma um pouco tensa e mole. O que isto permite são regiões no genoma, que estão bastante distantes de uma perspectiva linear, aproximando-se e interagindo de perto. Este adágio foi grosseiramente rejeitado pelo atual conjunto de ferramentas de enriquecimento (mapeamento) e, portanto, os resultados gerados são desproporcionais.
As regiões do genoma fazem parte de "grupos de ação" maiores ou caminhos that are technically series of chemical reactions accounting for a phenotype; healthy or diseased. When a diseased state is examined, the investigators are on the lookout for the biomarkers that have potentially gone awry and have apparently transformed the organismal body from toned para retorcida. Imagine perseguir uma doença difícil de combater com informações desalinhadas.
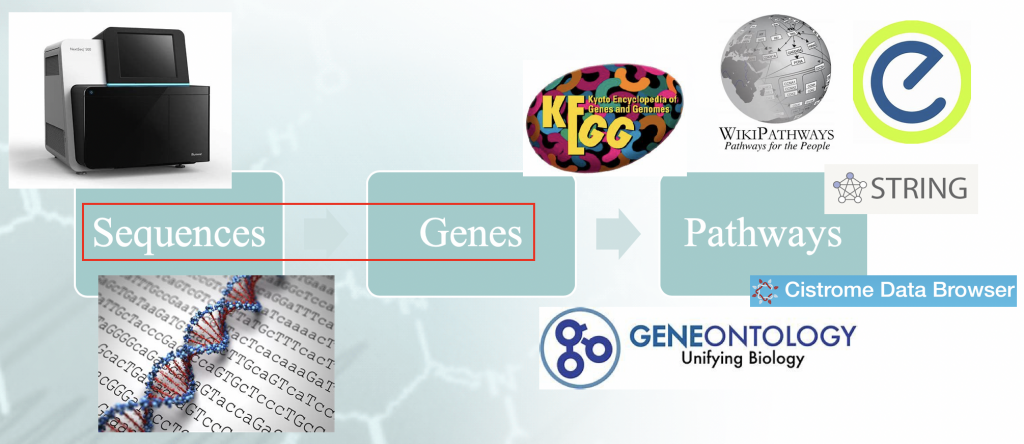 Declaração do problema - Fluxo de trabalho de análise de enriquecimento típico. Há uma certa
Declaração do problema - Fluxo de trabalho de análise de enriquecimento típico. Há uma certa
"idiomatismo" associado ao mapeamento das seqüências genômicas aos genes, e que orquestra o
resultados a jusante.
Organização do Genoma
Como aludido anteriormente, o genoma é suficientemente longo para ser armazenado linearmente dentro do núcleo de uma célula, para cada célula de nosso corpo ou qualquer outro organismo vivo, por esse motivo. Ao invés disso, esta entidade de 2 metros é esmagada e amontoada em uma estrutura aparentemente aleatória, com voltas em loop e redemoinhos como se pode imaginar. Este sem fim A adenina (A), citosina (C), timina (T) e guanina (G) estruturam topologias distintas dentro do núcleo enquanto se ajustam à exiguidade. Elas formam laços de cromatina, compartimentos/subcompartimentos, domínios/subdomínios que servem a um propósito, de acordo com o tipo de célula. (Note que diferentes tipos de células funcionam de forma diferente; uma célula nervosa tem outros negócios para atender do que uma célula muscular; cada uma tem um papel exclusivo a desempenhar).
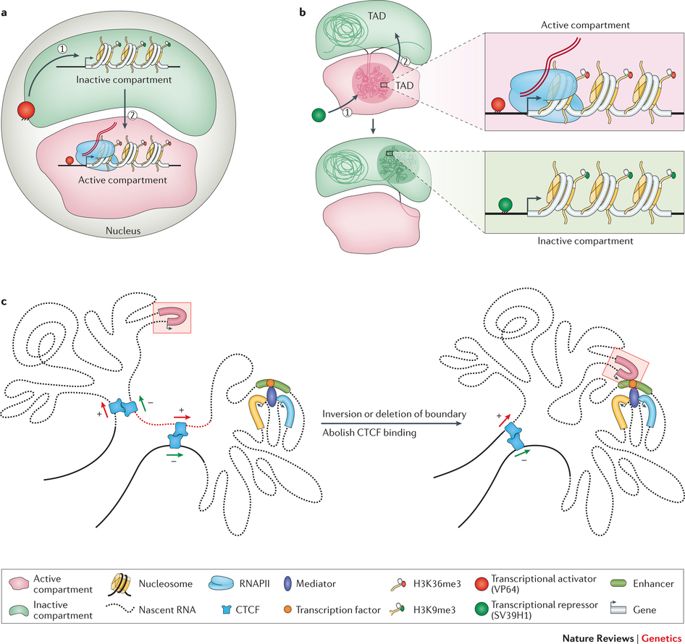
As bobinas do genoma e se espalham em espaços objetivos. (Crédito: https://doi.org/10.1038/nrg.
2016.112)
Bases de dados de caminhos
Existem diversas ontologias e bancos de dados, dos quais a Enciclopédia de Genes e Genomas de Kyoto (KEGG) (https://www.genome.jp/kegg/) e Gene Ontology (http://geneontology.org/As poucas ferramentas que vou apresentar na próxima seção (e que muitas vezes são opinantes) geram termos de enriquecimento que são "seletivamente" das referidas bases de dados. Com base em seus valores de significância estatística, deriva se eles realmente representam um fenótipo listado ou se são apenas uma questão de desenvolvimento aleatório (P.S. Há uma escrita sobre os valores p que presumivelmente ajudará os leigos a entender a idéia de significância estatística. Por favor, siga o link https://sway.office.com/WkyHrPnVB8Ec3zPD?play de acordo com sua conveniência).
Ferramentas de Enriquecimento
A análise de enriquecimento é um protocolo computacional de novo regiões genômicas a suas definições registradas nos bancos de dados que foram aludidos. Falando das ferramentas, (que atuam como um conduto), elas são classicamente estruturadas sob várias cabeças, a saber, análise de sobre-representação (ORA), pontuação de classe funcional (FCS), métodos baseados em topologia de caminho (PT) e métodos de interação em rede (NI).
Análise de Representação Excessiva
A análise de sobre-representação, através do dogma da distribuição hipergeométrica, avalia o conjunto de genes expressos diferentemente para aqueles que poderiam ser parte de um caminho biológico. Basicamente, um teste hipergeométrico considera quatro atributos para se chegar a uma decisão, a saber
- Número total de genes no ensaio considerado,
- Os genes expressos de forma diferente,
- Genes no caminho de destino a partir do número total de genes, e
- Genes expressos diferencialmente que ocorrem no caminho do alvo.
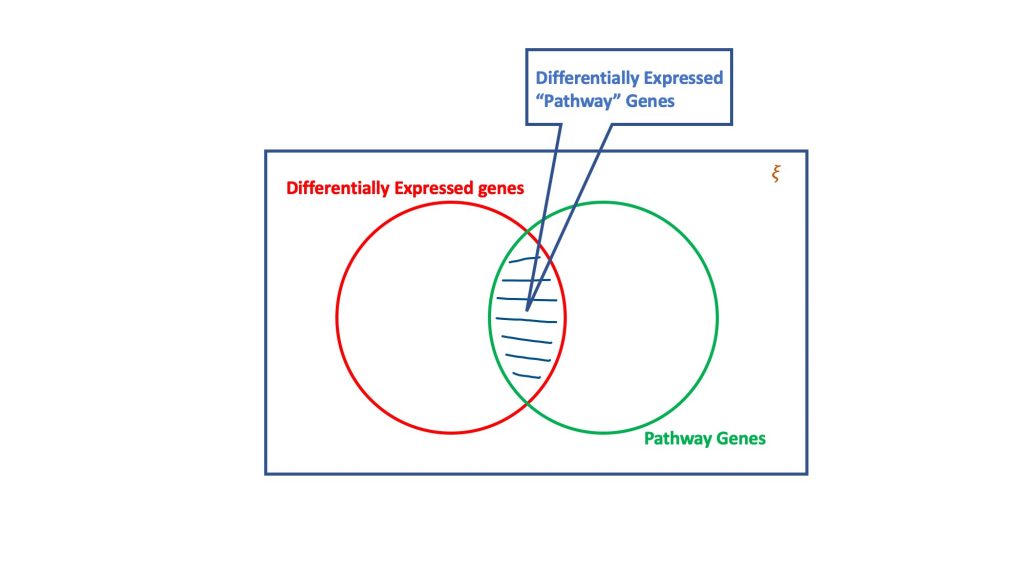
A essência do teste hipergeométrico
Despite being a simple and straightforward methodology, ORA presents its own limitations.
- Democracia em jogo; todos os genes são considerados igualmente; Por que isso é um problema? Vamos supor que os genes são filtrados com base em troca dobrável. Peneiramos genes que abraçaram diferenças de expressão maiores ou iguais a 2 vezes (dobras), tanto em sentido negativo quanto positivo. Embora o mínimo fosse 2 vezes, este fluxo de trabalho também capturaria genes com mudanças de expressão que fossem 3 vezes, 4 vezes e mais. Certamente, um gene com 4 vezes a disparidade de expressão é mais prudente do que um gene com 2 vezes a mudança. Esta manifestação é desconsiderada pela ORA.
- Considera apenas os genes mais significativosMais uma vez, consideremos um gene com troca dobrável 1,9999 ou p<0,0051113; geralmente, um p<0,05 é considerado estatisticamente significativo. A metodologia ORA glosa sobre este gene no resultado final. Claramente, há uma perda de informação e falta de flexibilidade. (P.S. Breitling et al. abordaram esta situação propondo uma extemporização para evitar limiares. A revisão emprega uma abordagem iterativa que acrescenta um gene de cada vez para compilar um conjunto de genes para os quais um caminho é o mais significativo possível).
- Nenhum gene funciona isoladamenteIsto decorre das limitações acima mencionadas que tratando o gene como uma entidade independente perde o nó da contribuição poligênica para um fenótipo. Um objetivo da análise da expressão gênica poderia ser elucidar coortes gênicas cujos padrões de expressão sejam congruentes. Esta sinfonia destaca genes funcionalmente semelhantes ou genes que trabalham em direção a um estado biológico comum.
- Caminhos mutuamente independentesA ORA também assume que os caminhos não funcionam em tandem (ou em sucessão). Isto é principalmente falho, pois uma série de reações químicas poderia muito bem preceder ou prosseguir uma à outra.
Pontuação da classe funcional
Contrary to ORA, FCS methods subsume all the contextual genes as well as their association estatísticas (fold-change, p-value) and compute a em funcionamento escore de enriquecimento para grupos de genes (baseado em alguns conhecimentos funcionais como Ontologia Genética ou caminhos do KEGG). ex. GSEA do Broad Institute (http://software.broadinstitute.org/gsea/index.jsp). Uma execução típica do FCS analisaria a mudança de expressão dos genes gerais na lista (não classificados por significância estatística ou outra coisa) de genes expressos de forma diferente em um experimento. O resultado primário da análise de enriquecimento do conjunto de genes é um escore de enriquecimento (ES) que reflete o grau em que um conjunto de genes está super-representado no topo ou na base de uma lista classificada de genes; por que topo e base? porque há os genes mais distantes do normal, em termos da mudança de expressão. Um resultado ES positivo para um conjunto de genes (ou um caminho de destino), GS) será indicativo dos genes da lista (GL) caindo no topo (a maioria não regulada; 1,2,3 ...), enquanto que uma pontuação negativa do ES significa que os genes componentes ficam na base (a maioria não regulada; n-3, n-2, n-1, n, onde n é o número total de genes). P.S. O ES se torna ES normalizado (NES) quando se corrige para o problema de testes múltiplos (taxa de falsas descobertas, por exemplo, o ES é o número total de genes). Método Bonferroni).
Em resumo, os métodos FCS são notavelmente melhores do que os métodos ORA por,
- evitar a exigência de um limiar arbitrário para classificar os genes como significativos ou não significativos.
- Apreciar informações sobre a expressão gênica para rastrear mudanças sistemáticas no caminho; isto torna a responsabilidade sobre a interdependência dos genes.
No entanto, os métodos da FCS também apresentam certas deficiências.
- Uma vez que os caminhos são analisados independentemente, os genes que regulam vários caminhos podem não ser contados.
- Muitos métodos da FCS classificam os genes em uma lista com base nas mudanças na expressão gênica. Um cenário em que a diferença de classificação reflete uma variação desigual (e possivelmente exponencial) na expressão poderia talvez ser uma medida injusta.
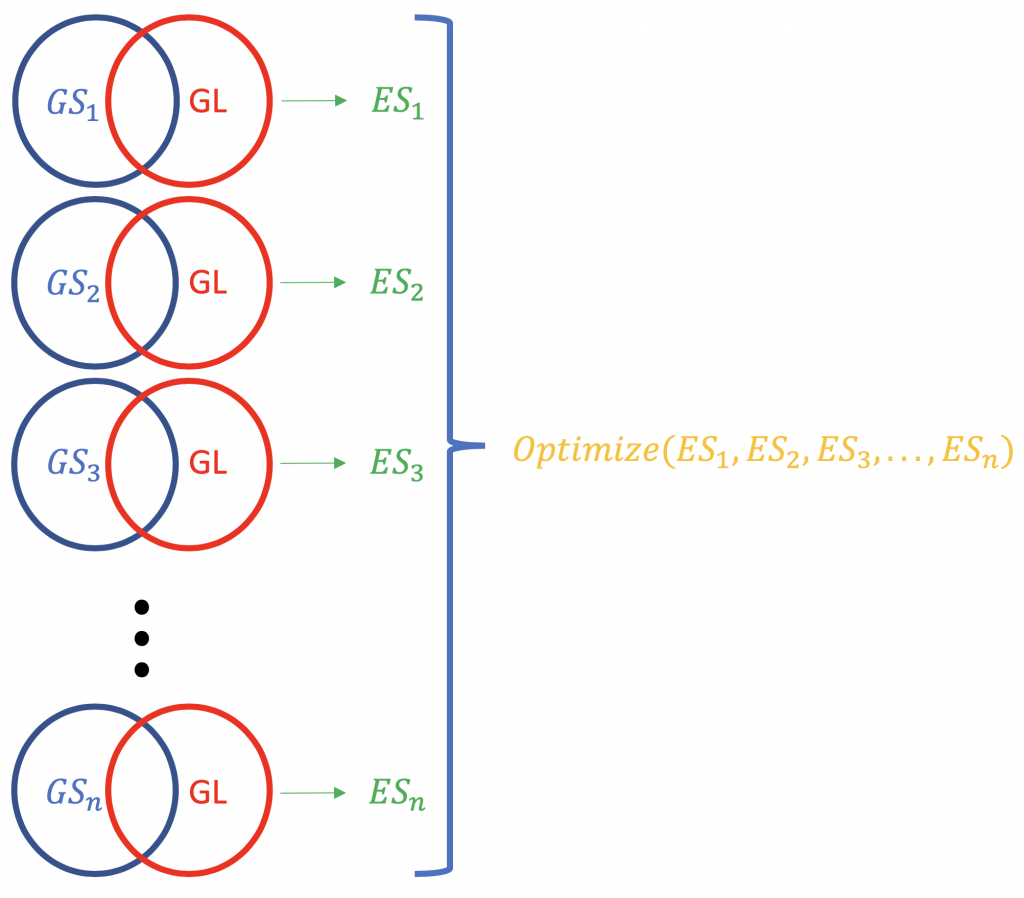
A normalização dos escores do ES relativos a uma lista de genes.
Abordagens baseadas na topologia do caminho
Uma privação crucial dos métodos ORA e FCS é que eles ignoram a estrutura dos caminhos. A ordem dos genes que são regulados em um caminho é essencial para traçar os efeitos causais. Compreensivelmente, pode haver exatamente dois caminhos com os mesmos componentes genéticos, mas a hierarquia de ativação pode ser totalmente diferente. Se fosse pelos métodos ORA/FCS, eles teriam resultado em termos de enriquecimento semelhantes. Isso é um problema. Os métodos de Topologia de Caminhos (PT) assumem exclusividade de função dependendo das interações específicas, o que também está de acordo com a lógica geral. Exemplos de ferramentas são SPIA (https://www.bioconductor.org/packages/release/bioc/manuals/SPIA/man/SPIA.pdf), GGEA, e PARADIGM. Geralmente, as ferramentas desta categoria devem ter uma pontuação local e uma pontuação global. O escore local no nível do gene deve calibrar as mudanças na expressão do gene e dos genes a montante, enquanto o escore global deve medir o nível do caminho para a relação com o conjunto de genes. No entanto, isto também faz com que os métodos PT se sobreponham aos dados para uma condição peculiar/tipo de célula.
Análise baseada na interação da rede
Esta é uma categoria bastante subestimada que ainda é pouco implementada, apesar de sua formulação datada. Métodos como EnrichNet , NetPEA (http://www.dx.doi.org/10.1109/BIBM.2013.6732493) have been proposed close to a decade ago, but they haven’t gained much traction because of limited tools available. This facade constraints the dexterity of the theme as no improvements are documented. That turns out to be an open-ended pesquisa problem.
O aparente Panacea
Você provavelmente tem agora uma narrativa sobre o enriquecimento fundamental/análise do caminho e o tipo de ferramentas que o ajudam. No entanto, como mencionei antes, todas as ferramentas disponíveis (que se enquadram nas categorias marcadas) dependem de um parâmetro inclinado de uma janela linear através da região consultada. Se os segmentos intervenientes do genoma caírem neste quadro, eles são listados como enriquecidosenão, não. O verdadeiro negócio é ter uma ferramenta que, talvez, introduza um centro e diâmetro de um círculo hipotético, como se fosse para destacar as interações genômicas baseadas na organização 3D para a região.

GREAT oferece várias opções avançadas para especificar a dimensão linear em torno do Site de Início de Transcrição de um gene.
 Enrichr apresenta uma seleção direta do tipo de genoma e do número de genes na suposta região linear.
Enrichr apresenta uma seleção direta do tipo de genoma e do número de genes na suposta região linear.
Como se deduz da Figura 4, não há apreciação pela "verdadeira" organização espacial do genoma. Este é um problema e uma profunda linha de falha que persiste no domínio atual da análise de enriquecimento. Não obstante, há também uma forte relevância de fábricas de transcriçãoA empresa é identificada como locais no espaço nuclear que atraem elementos reguladores distantes para "festejar internamente". Em tom de brincadeira, eu costumo dizer que, como alguém que está lívido com alguém ou alguma coisa costuma dizer, as fábricas de transcrição (personificadas) poderiam repreender o genoma...".A transcrição será feita por cima do meu cadáver, de qualquer outra forma!“. O tema adjacente das fábricas de transcrição é um tema de algumas discussões adjacentes e futuras. Entretanto, ele materializa o dogma das interações cis-regulatórias que é inadequado na prática contemporânea.
Para finalizar, gostaria de observar que a análise do caminho é uma fração crucial, e muitas vezes negligenciada, do Bioinformática gasoduto. Há sempre um espaço para dimensionar as metodologias existentes em conjunto com os dados genômicos, que estão evoluindo enquanto falamos. Quando o volume escalonado de dados se tornar disponível para nós, não será apenas um problema de infra-estrutura, mas também uma limitação algorítmica.
Necessidade de contratar um consultor em bioinformática? Trabalhar com cientistas freelance em Kolabtree. É grátis para postar seu projeto e obter citações de especialistas.
Peritos relacionados:
Profissional liberal da bioinformática | Genética vegetal | Biologia do Desenvolvimento | Terapia genética | Células-tronco |
Análise de dados de seqüenciamento de DNA |Genética animal | Interações medicamentosas | Genética e Genômica
Kolabtree helps businesses worldwide hire freelance scientists and industry experts on demand. Our freelancers have helped companies publish research papers, develop products, analyze data, and more. It only takes a minute to tell us what you need done and get quotes from experts for free.

Unlock Corporate Benefits
• Secure Payment Assistance
• Onboarding Support
• Dedicated Account Manager
Sign up with your professional email to avail special advances offered against purchase orders, seamless multi-channel payments, and extended support for agreements.


