



計算機生物学者 とKolabtreeのフリーランサー ショーリヤ・ジャウハリ writes about current challenges involved in conducting pathway analysis in bioinformatics and possible solutions to the problem.
In the contrasting era of “Imprecise Medicine” (propelling us to tune towards Precision Medicine) and inundation of biomedical data brought forth by advancements in instrumentation technologies, a lacuna persists that is largely premised over mapping data to information. The clinical experiments engender バイオマーカー (technically list of genes or genomic regions more contemporarily) that have to be expounded for their biological implications. The current suite of tools that facilitate such an endeavor is less purposeful as it neglects the いっそのこと真核細胞の核の中に収容されているゲノムの空間的な相互作用を明らかにする。この解説では、問題の本質を明らかにし、ゲノムの構成に光を当て、現在のマッピングツールを簡単に振り返り、可能な解決策を推測します。
悪魔は細部に宿る
ゲノムデータの解像度を高めるための数多くの努力は、重要な情報のスキームを下敷きにしています。私たちは、$100,000の解析はともかく、$1000のゲノムの可能性を心配しています。典型的な生物学的研究の下で、実験結果や症例の注釈を保持するレパートリーの大要が存在します。ある遺伝子の生物学的な意味合いや、その遺伝子が含まれる経路が病気と関連していることを示す定義があるかもしれません。繰り返しになりますが、このような動的な情報を持つデータベースは、以前は手作業で管理されていましたが、現在ではコンピュータやICTを駆使した自動化されたパイプラインによって知識管理が行われています。これらのデータベースは、コンセンサスを重視した科学的な知恵によって更新され、開始以来、何度も改訂されてきました。実験結果を生物学的な意味合いに結びつける導線は著しく弱くなっていますが、これは主に、根底にある「真の」生物学が否定されているためです。
私たちの体を構成する細胞のひとつひとつの核の中には、平均2メートルの長さのゲノムが収められています。細胞の大きさ、特に核の大きさが小さいため、ゲノムはやや窮屈な形で詰め込まれています。そのため、直線的な視点では遠く離れたゲノムの領域が近くに来て、相互に影響し合うことになります。この言葉は、現在の一連のエンリッチメント(マッピング)ツールでは大きく否定されており、そのため得られる結果も不均衡なものとなっています。
ゲノム内の領域は、より大きな「アクショングループ」の一部であったり パスウェイ that are technically series of chemical reactions accounting for a phenotype; healthy or diseased. When a diseased state is examined, the investigators are on the lookout for the biomarkers that have potentially gone awry and have apparently transformed the organismal body from トーン にしています。 トゥイッチ.ずれた情報で苦労した病気を追いかけることを想像してみてください。
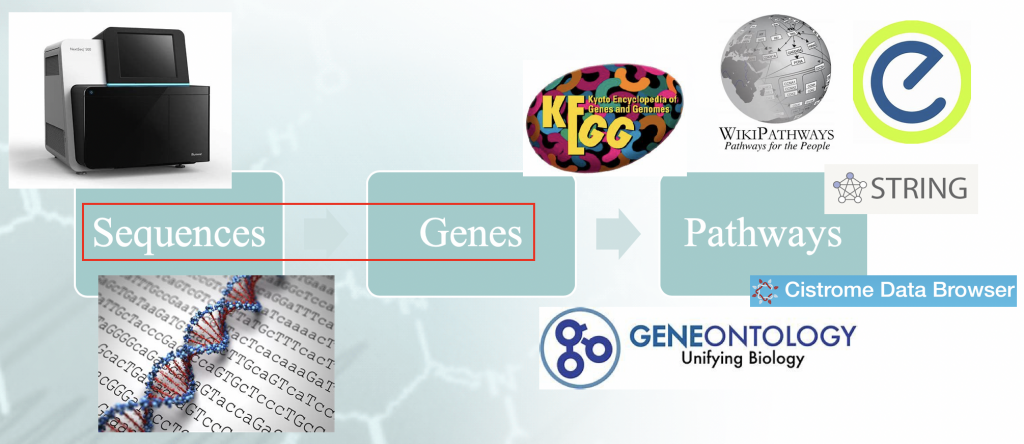 問題の提起 - 典型的なエンリッチメント分析のワークフロー。そこには、ある
問題の提起 - 典型的なエンリッチメント分析のワークフロー。そこには、ある
ゲノム配列の遺伝子へのマッピングに関連した「イディオマティズム」と呼ばれるもので、これによって
ダウンストリームの結果
ゲノム組織
前述したように、ゲノムは、私たちの体や他の生物のすべての細胞の細胞核の中に直線的に格納できるほど長いものです。しかし、この2メートルの物体は、一見すると無秩序な構造に押し込められており、想像するようなループやターン、渦巻きがあります。これは エンドレス アデニン(A)、シトシン(C)、チミン(T)、グアニン(G)という塩基対の文字列が、核内で異なるトポロジーを形成しながら、外見上の変化に対応していること。これらは、細胞の種類に応じて、クロマチンループ、コンパートメント/サブコンパートメント、ドメイン/サブドメインを形成し、目的を果たしている。(神経細胞は筋肉細胞とは別の仕事をしており、それぞれが専用の役割を持っている)。
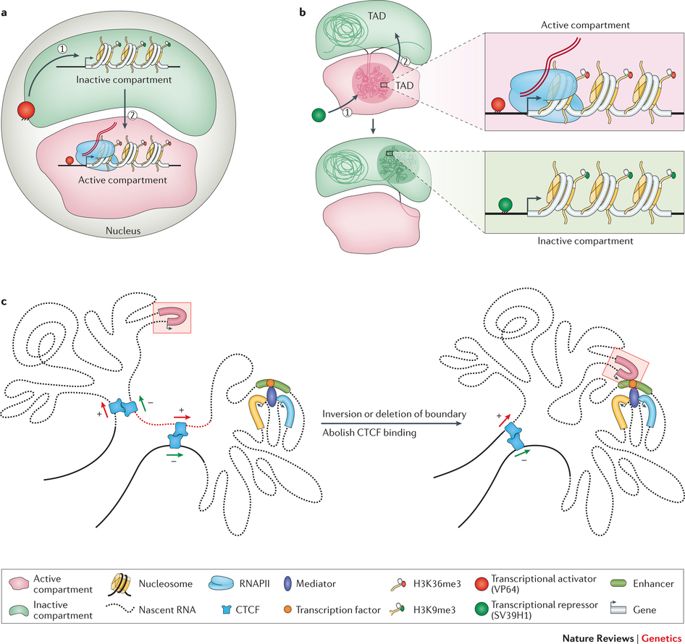
ゲノムはコイル状になっていて、目的の空間に広がっている。(クレジット: https://doi.org/10.1038/nrg.
2016.112)
パスウェイデータベース
様々なオントロジーやデータベースがありますが、その中でもKyoto Encyclopedia of Genes and Genomes (KEGG) (https://www.genome.jp/kegg/)とGene Ontology(http://geneontology.org/次のセクションで説明するいくつかのツール(そして多くの場合、意見が分かれている)は、上記のデータベースから「選択的に」エンリッチメント用語を生み出します。それらの統計的有意性の値に基づいて、それらが本当にリストアップされた表現型を表しているのか、それとも単なるランダムな展開の問題なのかが導き出されるのです。 統計的有意性.以下のリンクをご参照ください。 https://sway.office.com/WkyHrPnVB8Ec3zPD?play をご利用いただけます。)
エンリッチメントツール
エンリッチメント分析は、計算プロトコルの ノボ ゲノム領域を、これまで言及してきたデータベースに記録されている定義に照らし合わせます。そのツールとは、過剰発現解析(ORA)、機能クラススコアリング(FCS)、経路トポロジー(PT)に基づく方法、ネットワーク相互作用(NI)に基づく方法などである。
過剰表現の分析
過剰発現解析では、超幾何分布のドグマを介して、生物学的経路の一部である可能性のある差動発現遺伝子のセットを評価します。基本的に、超幾何学的検定では、以下の4つの属性を考慮して判定を行います。
- 考慮されたアッセイにおける遺伝子の合計数。
- 発現量の異なる遺伝子を
- 全遺伝子数のうち、ターゲットとなるパスウェイに含まれる遺伝子、および
- ターゲットパスウェイで発生する差動的に発現する遺伝子。
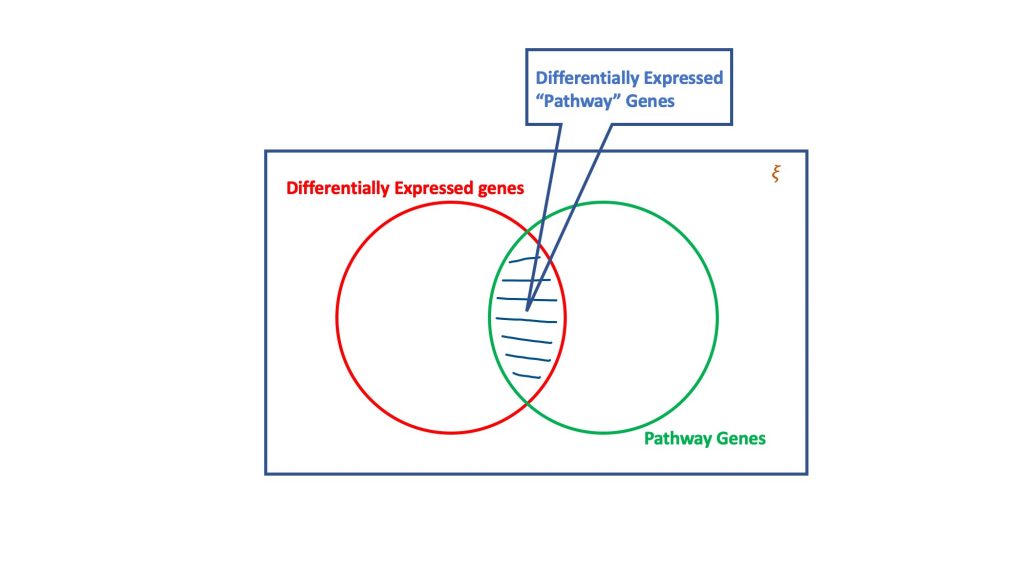
超幾何学的検定の本質
Despite being a simple and straightforward methodology, ORA presents its own limitations.
- 民主主義の実現、すべての遺伝子が平等に考慮されるなぜそれが問題なのか?仮に、遺伝子が以下の基準でフィルタリングされているとしましょう。 フォールドチェンジ.ネガティブ、ポジティブ両方の方向で、2倍以上の発現差を持つ遺伝子を選別します。最低でも2倍としたが、3倍、4倍、それ以上の発現変化がある遺伝子も対象とする。確かに、発現量が2倍の遺伝子よりも4倍の差がある遺伝子の方が慎重になる。この顕在化は、ORAでは割引されている。
- 最も重要な遺伝子のみを考慮するここでも、fold-changeが1.9999、p値が0.0051113の遺伝子を考えてみましょう。一般的には、p値が0.05未満であれば統計的に有意であると考えられます。ORAの手法では、最終結果でこの遺伝子を無視しています。明らかに、情報の損失と柔軟性の欠如があります。(P.S. Breitlingらは、閾値を避けるための外挿法を提案することで、この苦境に対処している。この修正では、一度に1つの遺伝子を追加して、パスウェイが最適に重要である遺伝子のセットをコンパイルする反復的なアプローチを採用している)。
- 孤立して働く遺伝子はないこのように、遺伝子を独立した存在として扱うと、表現型に対する多遺伝子の貢献の本質を見失ってしまうという、前述の限界があります。 遺伝子発現解析の目的の一つは、発現パターンが一致している遺伝子コホートを解明することである。このシンフォニーは、機能的に類似した遺伝子や、共通の生物学的状態に向かっている遺伝子を強調するものである。
- 相互に独立したパスウェイORAは、パスウェイが連続して機能しないことも想定しています。これは、一連の化学反応が互いに先行したり進行したりする可能性があるという点で、大きな欠陥があります。
機能クラスのスコアリング
Contrary to ORA, FCS methods subsume all the contextual genes as well as their association statistics (fold-change, p-value) and compute a ランニング 遺伝子グループのエンリッチメントスコア(Gene OntologyやKEGGパスウェイのような機能的知識に基づいている)。http://software.broadinstitute.org/gsea/index.jsp).典型的なFCSの実行では、実験で異なる発現をした遺伝子のリスト(統計的有意性などによるランキングではない)に含まれる全体の遺伝子の発現変化を分析する。遺伝子セットエンリッチメント解析の主な結果はエンリッチメントスコア(ES)であり、ある遺伝子セットがランク付けされた遺伝子リストの上部または下部に過剰に存在する度合いを反映している。ESスコアが正しければ、遺伝子セット(またはターゲットパスウェイ。 GS)は、リストの遺伝子を示すことになります(GLES スコアがマイナスの場合は、構成遺伝子が下位に位置していることを意味する(n-3, n-2, n-1, n, n, ここで n は遺伝子の総数)。P.S. ESはmultiple testingの問題(false discovery rate、例)を補正するとnormalised ES(NES)となる。 ボンフェローニ法).
要約すると、FCSの手法は、以下の点でORAの手法よりも優れているということになります。
- 遺伝子を有意か非有意かに分類するための恣意的な閾値の必要性を排除しています。
- パスウェイの系統的な変化を追跡するために、遺伝子の発現に関する情報を評価することで、遺伝子の相互依存性に関する説明責任を果たすことができます。
しかし、FCSの手法にもいくつかの欠点があります。
- パスウェイは独立して解析されるため、複数のパスウェイを制御する遺伝子がカウントされない場合があります。
- 多くの FCS 法は、遺伝子発現の変化に基づいてリスト内の遺伝子をランク付けする。発現量の不均等(指数関数的かもしれない)なばらつきをランクの差に反映させるというシナリオは、おそらく不公平な測定になるだろう。
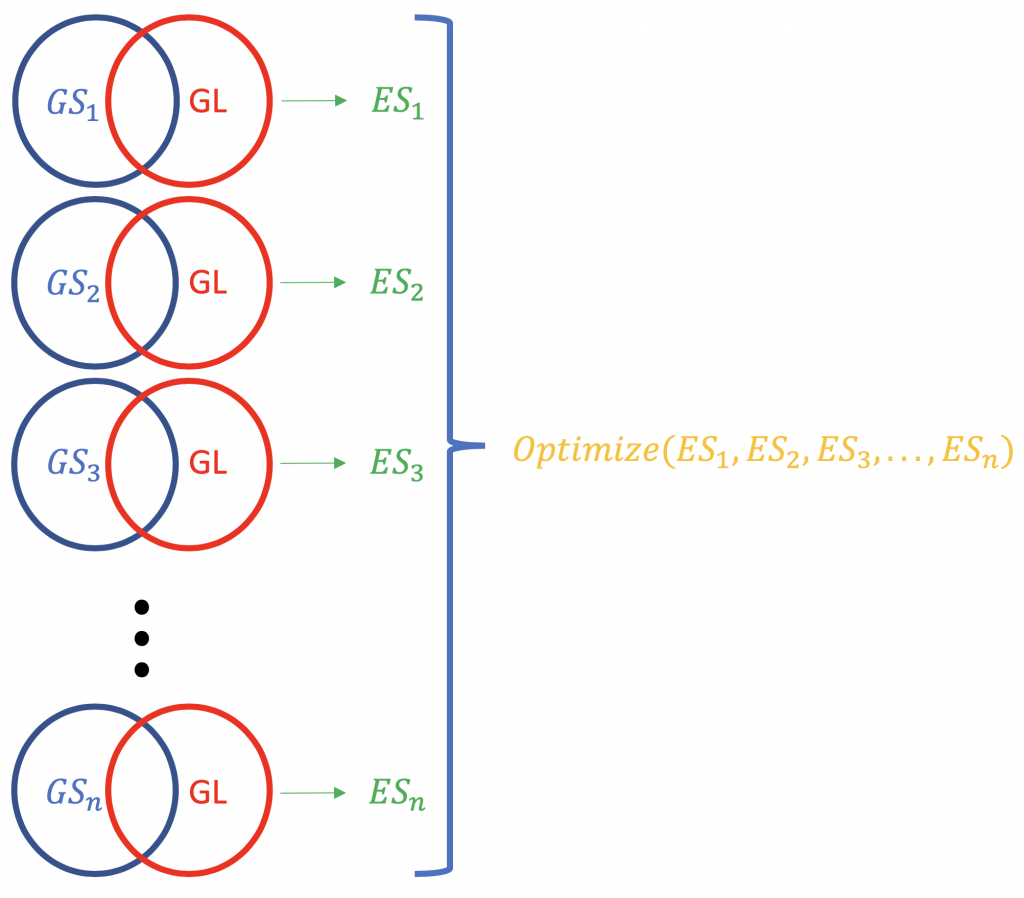
遺伝子リストに関連するESスコアを正規化する。
パスウェイ・トポロジーに基づくアプローチ
ORAやFCSの手法では、パスウェイの構造を無視していることが決定的な弱点です。パスウェイで制御されている遺伝子の順番は、因果関係を追跡するのに不可欠です。当然のことながら、同じ遺伝子成分を持つパスウェイが全く2つ存在しても、活性化の階層は全く異なる可能性があります。ORA/FCSの手法であれば、似たような濃縮項になっていたはずです。それが問題なのです。Pathway Topology(PT)法は、特定の相互作用によって機能が排他的になることを前提としていますが、これは一般的な論理にも合致しています。ツールの例としては、SPIA (https://www.bioconductor.org/packages/release/bioc/manuals/SPIA/man/SPIA.pdf)、GGEA、PARADIGMなどがある。一般に、このカテゴリーのツールは、ローカルスコアとグローバルスコアを持つ。遺伝子レベルのローカルスコアは、その遺伝子と上流の遺伝子の発現量の倍数変化を校正し、グローバルスコアは、遺伝子セットとの関連性を示すパスウェイレベルの指標となる。しかし、PT法では、特定の状態や細胞タイプのデータをオーバーフィットさせてしまうという問題がある。
ネットワーク・インタラクションに基づく分析
これは、古くからある手法にもかかわらず、まだほとんど実施されていない控えめなカテゴリーです。EnrichNet , NetPEA ()のような手法があります。http://www.dx.doi.org/10.1109/BIBM.2013.6732493) have been proposed close to a decade ago, but they haven’t gained much traction because of limited tools available. This facade constraints the dexterity of the theme as no improvements are documented. That turns out to be an open-ended リサーチ problem.
見かけ上は 万能薬
ファンダメンタル・エンリッチメント/パスウェイ解析と、それを支援するツールの種類については、もうお分かりいただけたと思います。しかし、先に述べたように、利用可能なすべてのツール(マークされたカテゴリーに属するもの)は、質問された領域を横切る線形の窓の歪んだパラメータに依存しています。ゲノムの介在するセグメントがこのフレームに入る場合、それらは以下のようにリストされます。 エンリッチドそうでない場合もあります。本当の意味でのツールは、おそらく、入力したものを センター そして 直径 仮定の円の中で、その領域のゲノムの相互作用に基づく3次元組織を強調するように。

GREATには、遺伝子の転写開始点を中心とした直線的な寸法を指定するためのいくつかのオプションが用意されています。
 Enrichrでは、ゲノムの種類と推定される線形領域の遺伝子数を簡単に選択できます。
Enrichrでは、ゲノムの種類と推定される線形領域の遺伝子数を簡単に選択できます。
図4から推測されるように、ゲノムの「真の」空間的構成については評価されていません。これは、現在のエンリッチメント解析の領域に残っている問題であり、深い断層でもあります。それにもかかわらず、次のような重大な関連性もあります。 転写工場は、離れた場所にある制御因子を「社内パーティー」に誘う核空間のサイトとして同定されました。冗談めかしてよく言うのですが、誰かや何かに腹を立てている人が普通に言うように、転写工場(擬人化)がゲノムを叱責することは考えられます。書き取りは私の死体の上で行われますが、他にはありません".転写ファクトリーという隣接したテーマは、いくつかの隣接した将来の議論の対象となります。しかし、このテーマは、現代の実務において問題となっているシス・レギュレーター相互作用のドグマを具体化したものである。
最後に、パスウェイ解析は非常に重要でありながら無視されがちな部分であることを指摘したいと思います。 バイオインフォマティクス パイプライン進化し続けるゲノムデータに合わせて、既存の方法論を拡張する余地は常にあります。膨大な量のデータが得られるようになると、インフラの問題だけではなく、アルゴリズムの問題も発生してきます。
が必要です。 バイオインフォマティクスのコンサルタントを雇う?Kolabtreeでフリーランスの科学者と仕事をしてみませんか?無料でプロジェクトを投稿し、専門家から見積もりをもらうことができます。
関連する専門家
バイオインフォマティクス・フリーランサー | 植物遺伝学 | 発生生物学 | 遺伝子治療 | 幹細胞 |
DNAシークエンスデータの解析 |動物遺伝学 | 薬物相互作用 | 遺伝学・ゲノミクス
Kolabtree helps businesses worldwide hire freelance scientists and industry experts on demand. Our freelancers have helped companies publish research papers, develop products, analyze data, and more. It only takes a minute to tell us what you need done and get quotes from experts for free.

Unlock Corporate Benefits
• Secure Payment Assistance
• Onboarding Support
• Dedicated Account Manager
Sign up with your professional email to avail special advances offered against purchase orders, seamless multi-channel payments, and extended support for agreements.


