



An exhaustive guide to 規制遵守 for IVD manufacturers, written by Sundeep Agarwal経験豊富な IVDRコンサルタント.
IVDRとは?
The European Commission’s (EC) In Vitro Diagnostic Regulation (EU IVDR 2017/746) is a ‘legislative framework’ and a way forward towards global IVD safety, which assures that only reliable and effective IVDs are in the market. The European Commission is trying its best to make the ヘルスケア system safer and error free in terms of diagnosis or outcomes.
体外診断用医療機器について (IVDDは98/79/ECが指令であるのに対し、IVDRは欧州経済領域(EEA)内の製造業者、輸入業者、ユーザー、公認機関、国家当局、および欧州以外の製造業者、サプライヤーが欧州市場でIVDを販売する、または販売しようとするすべてのEOに適用される法令(規則)である。
IVDRは113条(10章)、15付属書から構成されているのに対し、IVDDは24条、10付属書から構成されています。IVDRは長大で厳しい規制であることは間違いないが、規制の変更や要求事項がより透明化されていることが良い点である。
ライフサイクルに基づくアプローチを重視している。2022年5月26日から適用され、経済事業者(EU域外のメーカーを含む)は、その計画・実施に向けて積極的に準備することが期待されています。このプロセスにおけるすべての関係者は、欧州経済領域(EEA)の体外診断用医薬品市場に対して等しく責任を負うことになります。
- まず、EU IVDRに関するトレーニングプログラム(オンラインまたはオンサイト)を実施し、組織内の全員が必要な変更点を認識できるようにすることが重要です。
- このプロセスや義務について、すべてのサプライヤー、下請け業者、サービス提供者に公式のコミュニケーショ ンを行う必要があります。
- EU IVDRで要求される技術文書を更新するために、彼らのリソース、有能なチームの可用性をチェックするためにギャップアセスメントを実行する。ISO 13485: 2016の認証を取得していると、コンプライアンスを確立する上でさらに有利となる。
- 一針入れば九死に一生を得る」ため、移行のごく初期段階から主題専門家や外部コンサルタントを雇うことが望ましい(必要な場合)。
- 本ブログでは、EU IVDR 2017/746に基づく様々な条文や附属書に記載されているノーティファイドボディや所轄官庁による期待に応えるための詳細な概要と実践的なヒントを紹介します。
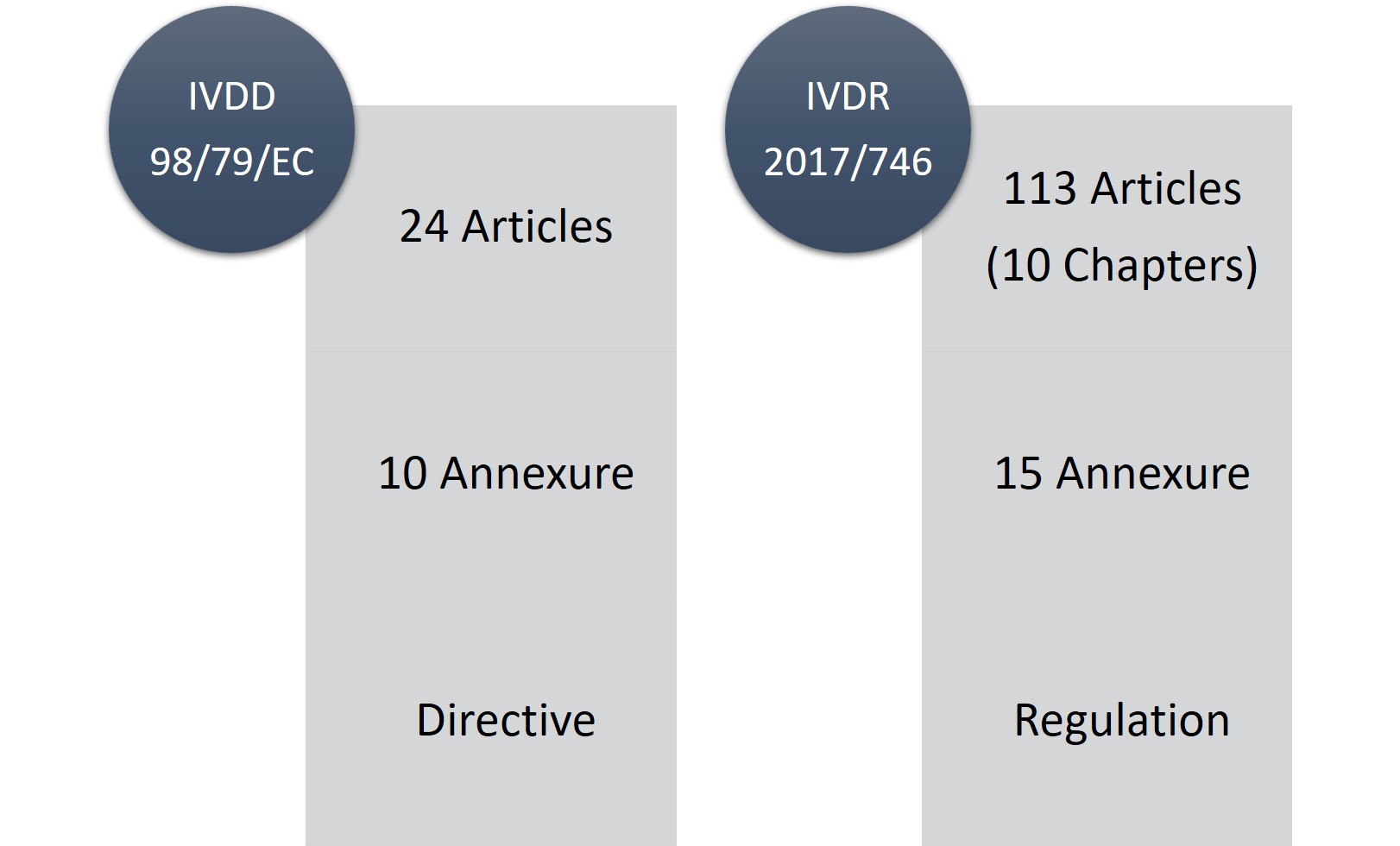
図1: IVDDとIVDRの比較
1.IVDR対応と商流変更への準備
企業にとって最も重要な経営判断は、自社のIVDを引き続き欧州経済領域(EEA)に置くかどうかの結論を出すことでしょう。その答えが「はい」であれば、できるだけ早くNBから見積もり(コスト)、スケジュール、審査範囲、製品コードなどを入手する必要があります。指令から規制への移行により、安全性と有効性を立証し、CE認証を取得するために、コンプライアンスと強固な技術文書が要求されます。 IVDRは、臨床エビデンスに大きく依存しています。 すなわち、科学的妥当性、分析的性能、臨床的性能。 安全性と有効性を確立するために
CE認証のプロセスにノーティファイド・ボディ(NB)が関与することは、この規制の顕著な特徴である。これはまた、経済事業者にとっての追加投資を意味し、間接的に製品コストを増加させる可能性があります。
の任命がありました。 "規制遵守責任者(PRRC)" in accordance with Article 15 of EU IVDR 2017/746 is now mandatory; who shall assure the conformity of QMS, declaration of conformity, technical documentation, post market surveillance and reporting of adverse events are in compliance to EU IVDR.Manufacturers should ensure that the entire transition (including new certification application) is completed before the expiry of their existing IVDD Certificate or Self-certified Declaration of conformity. Certificates issued by notified bodies in accordance with IVDD 98/79/EC from 25 May 2017 shall become invalid after 27 May 2024. Be aware of the new timeline for application as per the EC official press release [1] dtd.20th 2022年12月
2.分類の明確な理解
IVDRのAnnex VIIIに基づく新しい分類ルールを再考し、以前の分類に影響を及ぼしていないかどうかを確認する。
CE認証プロセスの準備の前に、正しい分類を行うことが不可欠です。それができない限り、適合ルートが不明確になり、IVDR要求事項への適合を遅らせたり、無効にしたりすることになる。
IVDRは リスクベースアプローチ は、ノーティファイドボディと所轄官庁の管理が強化されたデバイスを分類するために使用されます。この規則では フォーリスククラス:クラスA(最低リスク)、クラスB、クラスC、クラスD(最高リスク)。 7 分類規則 を正しく分類するために必要です。IVDR の特徴は、ソフトウェアも附属書 VIII の実施規則 1.4 で分類されていることである。「機器を駆動する、または機器の使用に影響を与えるソフトウェアは、機器と同じクラスに分類されなければならない。ソフトウェアが他の機器から独立している場合、それ自体で分類されるものとする。[2]".これは、IVDR で規制されるソフトウェアの範囲を示すものである。また、製造者はそれに応じてソフトウェアのベリファイとバリデーション(附属書 II の 6.4)を行う必要がある。
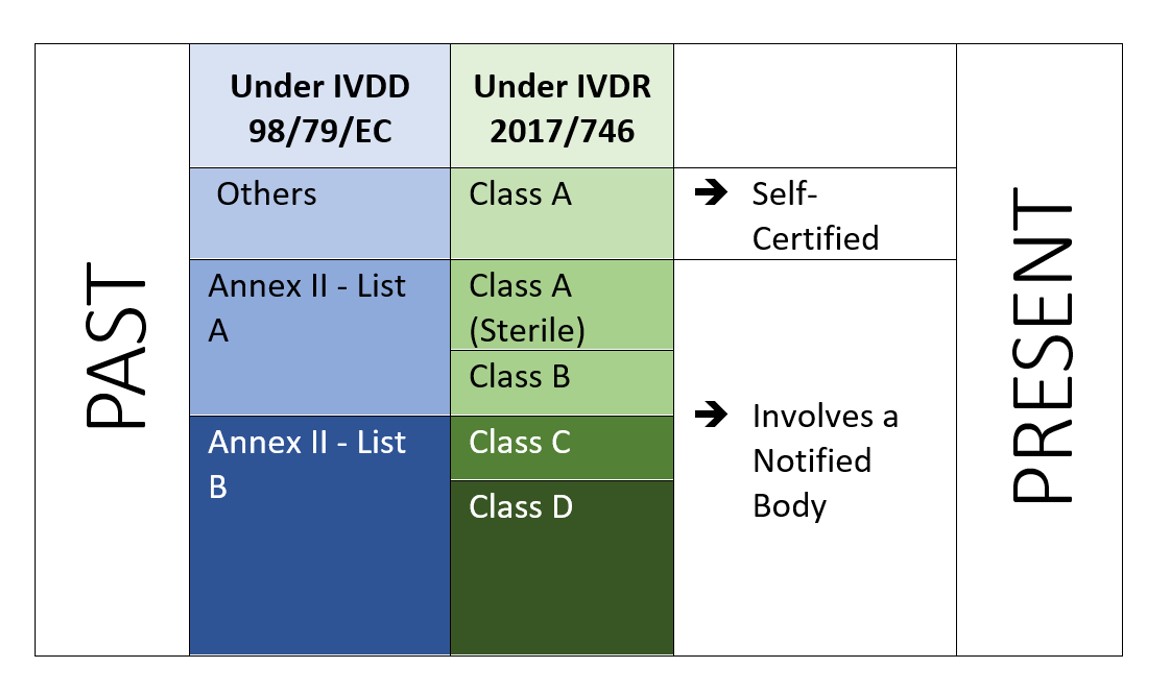
図2:IVDR 2107/746に基づくリスクベースの分類
3.届出機関の関与
ノーティファイドボディ(NB)の役割は中核的な要素のひとつであり、したがって、従来の「自己証明」の方法に対して、より多くのメーカーがノーティファイドボディの監査と認証を受ける必要が出てくることになる。経済事業者は、適合性評価ルート(EU IVDR の Annex IX, X, XI)を慎重に決定する必要があります。[3]).
IVDRでは、追加投資が必要なだけでなく、技術文書や品質管理システムがIVDRの新しい要求事項を満たしていることを保証しなければならない。IVDDでは、ほとんどのIVDが自己認証(92%)であり、ノーティファイドボディの関与は必要ありません(市場に流通しているIVDのうち8%を除く)[4]。しかし、新しいIVDRの下では、このシナリオは同じではありません。
ある調査によると “The impact of the new European IVD-classification rules on the notified body involvement” by National Institute for 公衆衛生 and the Environment, Bilthoven (Netherlands) RIVM Letter report 2018-0082, A. van Drongelen et al., nearly 85% of all IVDs will be requiring Notified Body involvement, leaving only 15% of IVDs eligible for self-certification[5].
また、体外診断用医薬品(IVD)メーカーは、新しい分類と認証プロセスを遵守するために大きな変化を経験することになります。 さらに、機器の使用目的やリスククラスに応じて、メーカーは、自社製品を審査し、認証を取得できる可能性のある指定NBを特定する必要があります。最もリスクの高いIVD(クラスD)では、ノーティファイドボディ(NB)や所轄官庁(CA)の関与に加えて、EUリファレンスラボや専門家パネルによる性能主張の検証も必要となります。現在、EU IVDRに指定されているノーティファイドボディ(Notified Body)は6機関のみです。ノーティファイドボディが利用できないことによる予期せぬ遅延を避けるためにも、申請手続きの開始をお急ぎください。

図3:IVDRにおける指定届出機関一覧[6]。
4.品質マネジメントシステム(QMS)の構築
体外診断用医薬品製造業者は、堅牢で信頼性の高い品質管理システム(QMS)を施設内に構築することが期待されています。これは、IVDR第10条に基づく製造業者の一般的な義務である.品質マネジメントシステムは、様々な要求事項の中でも特に重要なものであり、これがなければ製造者は認可を受けることができない。
QMS is to ensure that manufacturing, change control, customer complaints, resource management, supplier &sub-contractors’ controls and validation, performance evaluation, quality test, UDI Labelling, Post market surveillance etc. are according to approved QMS and Post Market Surveillance (PMS) plans.
PRRCは、製造者が第10条の要件を満たし、プロセスにおいてノーティファイドボディ(NB)を必要としない場合、クラスA IVDを「自己認証」(附属書IVに基づく適合宣言書の発行)することを確認しなければならない。
5.サプライチェーンの混乱に備える
Throughout the world, manufacturer depends largely on their supply chain and raw material to produce and deliver IVDs that are safe, accurate, and effective forthe intended use. Hence regulatory and quality concerns are also evolving to a higher level when it comes to the suppliers and sub-contractors’ controls. Manufacturer are therefore expected to proactively communicate the supply chain about their obligations and responsibilities of the suppliers and subcontractors. Legal manufacturer shall demonstrate adequate supplier control and monitoring, assure the supply chain is in compliance to the regulatory aspects of IVDR, reconsider the need for data integrity and quality of supplier data, implement robust supplier risk management and performance monitoring and periodically audit the supplier based on the associated risk to the finished products. 規制当局やノーティファイドボディは、サプライヤー管理のレベルを明確に文書化し、サプライヤーが提供する製品やサービスのリスクを軽減する可能性があることを根拠を持って実証することを、合法的なメーカーに強調している。
6.監査・査察の準備の確保
IVDR 第 88 条「市場監視活動」によると、所轄官庁は、経済事業者、供給業者、下請業者の敷地内、 および必要に応じて専門ユーザーの施設に対して、公表(抜き打ち)検査を行うものとされています。製造者は、設計・製造情報の技術文書に、サプライヤーや下請け業者を含む、製造活動が行われるすべての拠点の特定に関する情報を含めなければならない。QMS 審査を行うノーティファイドボディ(NB)は、様々な製造拠点、及びそのサプライヤーや下請け業者間の連携と責任分担を明らかにしなければならない。この情報は、NB がサプライヤまたは下請け業者のいずれか、あるいは両方を審査する際に考慮され ます。 製造者の供給者の施設は、完成機器の適合性に重大な影響を与えると考えられる場合、基本的にNBの監査を受けなければならない(特に、製造者がその供給者を十分に管理していることを実証できない場合)。
7.抜き打ち監査への対応計画
認証取得後のモニタリングでは、NBは、製品試験を実施する製造業者及びその下請業者又は供給業者に対する抜き打ちの現地監査、並びに製造業者を拘束し認証決定に関連する条件(定められた間隔での臨床データの更新等)が遵守されていることのモニタリングを実施する。さらに、t通知機関は,製造者のサイト及び,適切な場合には製造者の供給者及び/又は下請業者のサイトについて, 少なくとも5年に1回,無作為に抜き打ち監査を実施しなければならず,これは定期サーベイランス審査と組み 合わせて実施することができる。
8. 市販後調査活動の強化
製造業者は、市販後サーベイランスの要件を強化し、警戒と市場サーベイランスについてEU加盟国間で調整するメカニズムを開発することを強く推奨される。クラスC及びクラスDの機器に適用されるサーベイランス評価(附属書IX)では、通知機関は少なくとも12ヶ月に一度、定期的に適切な監査と評価を実施しなければなりません。これには、製造者及び供給者及び/又は下請け業者の敷地内における監査が含まれるものとします。製造業者は、基本的に、事故及び現場安全是正処置(FSCA)の記録及び報告のための手順を開発しな ければならない。
9.ユニークデバイス識別子(UDI)&EUDAMED
製造者は、機器を識別し、トレーサビリティを促進するために、UDIのシステムを構築しなければならない。UDIを付与するシステムを運用するために、GS1、HIBCC、ICCBBA、IFA GmbHなどの認定された発行機関(IE)のリストを参照することができる。現在、これらのIEは27日から有効です。th 2019年6月ですが、その有効性を確認しつつ、最終的に導入を決定するのが賢明でしょう。
欧州医療機器データベース(EUDAMED)は、欧州連合で入手可能なすべての医療機器の概要を提供する予定です。以下の6つのモジュールで構成されています。
- 俳優登録。
- UDI(Unique Device Identification)と機器登録。
- ノーティファイドボディと認証書。
- 臨床試験と性能試験。
- 自警団とポストマーケットサーベイランス
- マーケットサーベイランス
包括的なEUDAMEDを通じて透明性を向上させるため、経済事業者の情報の一部は一般に公開される予定である。一方、機密情報は、経済事業者、スポンサー、EU加盟国の公認機関および所轄庁のみがアクセスすることができます。
10. "自社製品 "に対する要求事項
Health institution developing ‘in-house devices’ (or ‘laboratory-developed tests’) which are meant to be used by the same health institution shall not be marketed or sold to other legal entity. Such devices may be used for the diagnosis and treatment, especially for rare diseases. The institution is expected to comply with only the requirement of Annex I of IVDR (general safety and performance requirements), and exempted from rest of the regulation until 26 May 2024; provided the health institution meets a number of conditions set out in Article 5(5) of the Regulation and has an appropriate quality management system, which complies to the international standard setting out the quality and competence requirements for medical laboratories (EN ISO 15189) or other national provisions, and is able to justify that target patient group’s specific needs cannot appropriately be met by an equivalent device available on the market.
リファレンス
[1] EC公式プレスリリース(20日)th 2021年12月、体外診断用医療機器規制の段階的な展開。アクセスはこちら https://ec.europa.eu/commission/presscorner/detail/en/IP_21_6965 [2]REGULATION (EU) 2017/746 OF THE EUROPEAN PARLIAMENT AND OF THE COUNCIL of 5 April 2017 on in vitro diagnostic medical devices and repealing Directive 98/79/EC and Commission Decision 2010/227/EU; ANNEX. VIII 分類規則、1.実施規則 ポイント 1.4 Page 304 [3]体外診断用医療機器に関する2017年4月5日の欧州議会および理事会の規則(EU)2017/746、指令98/79/ECおよび委員会決定2010/227/EUを廃止する。ANNEX IX 品質マネジメントシステム及び技術文書の評価に基づく適合性評価, Page 306, ANNEX X 型式検査に基づく適合性評価, Page 314, ANNEX XI 生産品質保証に基づく適合性評価, Page 317.
[4] プレスリリース dtd.2021年10月14日、ブリュッセル;公衆衛生。欧州委員会、体外診断用医療機器に関する新規制の漸進的導入を提案 [5]欧州の新しいIVD分類規則がノーティファイドボディの関与に与える影響; : オランダで登録されたIVDに関する研究; van Drongelen A, de Bruijn A, Pennings J, van der Maaden T 英語版32p 2018, RIVM letter report 2018-0082 [6] 上記一覧は、2012年3月31日時点のデータに基づくものです。最新のリストは、ECのウェブサイトをご覧ください。 https://ec.europa.eu/growth/tools-databases/nando/index.cfm?fuseaction=directive.notifiedbody&dir_id=35Kolabtree helps businesses worldwide hire freelance scientists and industry experts on demand. Our freelancers have helped companies publish research papers, develop products, analyze data, and more. It only takes a minute to tell us what you need done and get quotes from experts for free.

Unlock Corporate Benefits
• Secure Payment Assistance
• Onboarding Support
• Dedicated Account Manager
Sign up with your professional email to avail special advances offered against purchase orders, seamless multi-channel payments, and extended support for agreements.


