



Sundeep Agarwal, 医療機器 regulatory consultant on Kolabtree, shares the essential requirements of a FDA 510k premarket notification to ensure success.
現在の $156 億円の市場規模があり、予想される $208 億円 [1] by 2023, the US medical device market is undoubtedly lucrative. Additionally, the aging population, history of chronic diseases, the ヘルスケア system and disruption in supply chain encourages global manufacturer to invest and expand their business horizon in the United States. Like any other global manufacturer, if you think you can leave an imprint in the world largest medical device market, you have just hit on the right blog to prepare and understand the regulatory requirement with respect to FDA’s 510(k)for your device which is utmost vital for the application process.
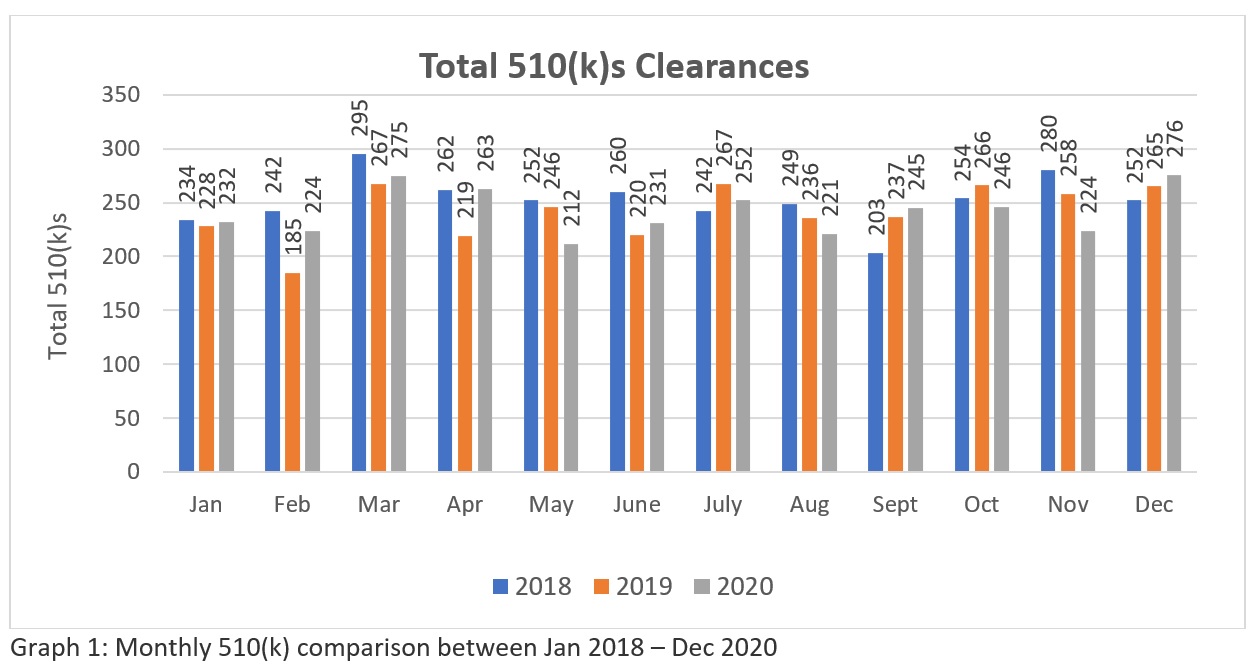
A trend analysis of three years FDA’s 510k clearances data (Source: https://www.fda.gov/medical-devices/device-approvals-denials-and-clearances/510k-clearances)によると、合計 30252018年1月から2018年12月までにクリアした-510(k)swere。 2894 - 2019年1月から2019年12月の間に510(k)sを取得し 2901 - 2020年1月から2020年12月の間にクリアされた510(k)s。これらの数字(表1参照)は、現在、FDAは、米国のヘルスケア市場のニーズを満たすために、毎月200件以上の510(k)を承認する可能性があることを示しています。一方、グラフ表示(グラフ1参照)は、世界的なパンデミックの中で、FDAの審査アプローチが一貫していることを示している。しかし、この数字は、世界的に行われている膨大な数の申請と比較すると、それほど大きなものではありません。今後、FDAがリソースを増やしてくれることを期待しています。そのため、メーカーは初回の申請で確実に通過できるよう、入念な準備と申請を行う必要があります。
| 年 2018年 | 年 2019年 | 2020年 | |
| 510(k)sの総数 | 3025 | 2894 | 2901 |
| サマリーとの合計 | 2885 | 2735 | 2759 |
| ステートメントとの合計 | 140 | 159 | 142 |
表1:2018年1月~2020年12月の間にクリアされた510(k)の年次を示す表です。
FDA 510kの要件。鍵を見つける
FDA(米国食品医薬品局)の基準に精通していないと 510(k)の準備と提出 のプロセスは、本当にストレスが多く、困難なものです。機器メーカーが、クラスI、II、IIIのいずれかに該当する機器を米国で販売しようとしている場合は、クラスI、II、IIIのいずれにも該当しません。 除外 510(k)規格に準拠したもの、または 必要ない 市販前承認申請(PMA)を行っていれば、510(K)を受けることができます。さらに進むと、メーカーは「プリディケート・デバイス」と「実質的同等性(SE)」という用語を明確にしておく必要があります。メーカーが同等性を主張したいと考えている米国で合法的に販売されている機器は、一般的にプレディケート機器として知られています。一方、実質的同等性とは、メーカーが米国市場に投入しようとする新製品が、特に同じ使用目的や技術的特性に関して、既存のプレディケート機器と同等の安全性と有効性を有するという証拠に基づく比較を確立することを意味します。新製品の技術的特性が異なる場合でも、安全性と有効性には違いがなく、メーカーはその製品が既存の合法的に販売されている製品と同等の安全性と有効性を有することを証明することができます。
プロセスの解明
510(k)申請を成功させるためには、製品コードの正確な分類、米国市場における先行製品の識別と入手可能性、十分に計画された実質的な同等性の証拠との比較、強固な品質管理システム、設計管理、現行のFDAフォームの厳格な遵守、およびRTA(Refuse to Accept)チェックリストの最適な利用などがあります(図1も参照)。RTA(Refuse to Accept)チェックリストは、FDAが510(k)の実質的な審査を行う際に従う様々な受け入れ基準を示しています。 3種類の510(k)すべてについて、異なるタイプのRTAが用意されています。RTAチェックリストは以下からアクセスできます。 これ.
メーカーは、ページ数や、試験結果や研究内容を提出するだけでは、評価されないことに注意する必要があります。 510(k)クリアランス むしろ、科学的な正当性と証拠に基づいて、(FDAがガイダンス文書で推奨しているように)十分に説明され、順序立てて書かれた文章が、一定の時間枠の中でFDAによって慎重に検討されることが、最終的に510(k)を達成することになります。510(k)の大半は、実質的な同等性を主張するための明確な証拠がない(従来型の510(k)の場合)、またはFDAが認めた適合性基準との比較が不十分である(略式の510(k)の場合)という理由で却下されています。また、設計管理を怠り、品質システムが確立されていないことも、このような失敗の大きな理由となっています。最後に、有能なチームが存在しないことや、規制に関する明確な理解がないことも、上記に加えて不合格の原因となる否定できない要因です。

図1:市販前通知510(k)の検討ポイント
FDA 510(k) 市販前通知の種類
一般的に、メーカーがFDAに提出する510(k)sには、3つのタイプがあります(図2参照)。それらは以下の通りです。
(1) トラディショナル 510(k) - 最も一般的で、510(k)の大半はこの申請タイプです。
(2) スペシャル 510(k) - 既に承認されている既存の機器において、ラベルやデザインの変更、または表示の一部変更が行われる場合にのみ必要となります。その内容は、21 CFR Part 807.87 および 21 Part 807.90 で定義された要件を満たす必要があります。
(3) 簡略化された 510(k) - FDAの承認した基準に基づいて、特別管理、ガイダンスの使用に関する報告書または適合宣言書を作成できる場合に適用されます。

図2:市販前通知510(k)の種類
510k申請プロセス
申請の前に、メーカーはFDAに組織を登録する必要があります。このプロセスは、21 CFR Part 807に基づく施設登録と呼ばれ、FDAに直接手数料を支払った後、毎年更新されます。2021年の会計年度では、施設登録の手数料は$ 5,546となっています。正確な料金は、FDAユーザーフィープログラムの公式サイトでご確認ください。
FDAは、従来型510(k)または短縮型510(k)において、20のセクションを推奨していますが、必ずしもすべてのセクションがメーカーに適用されるわけではありません。特定のセクションの情報が自社のデバイスに適用されない場合は、セクションの見出しを記載し、その下に「This section does not apply」または「N/A」と記載することがあります。FDAのガイダンス文書で推奨されている510(k)の主要セクションは以下の通りです。
- 医療機器ユーザーフィーカバーシート(Form FDA 3601)。 メーカーがFDAに支払ったユーザーフィーの受領を示す。
- Center for Devices and Radiological Health (CDRH)Premarket Review Submission Cover Sheet (Form FDA 3514)です。 これは、FDAに組織や提出物に関するあらゆる管理情報を提供するための任意のフォームです。
- 510(k)カバーレター 510(k)に関する目的、内容、管理情報に関する説明をこの手紙に盛り込む必要があります。Format for Traditional and Abbreviated 510(k)s Guidance for Industry and Food and Drug Administration Staff; dtd September 13, 2019」のAppendix Aを参照することが推奨される。
- Indications for Use Statement (Form FDA 3881)をご覧ください。 これは、510(k)全体で統一されるべきである。また、その機器が処方箋用として販売されるのか、OTC(Over-the-Counter)用として販売されるのかを定義する必要があります。
- 510(k)Summaryまたは510(k)Statement。 21 CFR part 807に基づいて作成すること。ここでは、メーカーが510(k)を要約し、残りの内容に関する情報を組み込むことが期待されています。
- 真実性と正確性の表明 これは、510(k)に関連してFDAに提出されたすべての情報が真実かつ正確であることを証明する組織の権限のある人による声明です。
- クラスIIIの概要と認証 クラスIII機器のみに適用される。安全性と有効性の要約であり、合理的な調査が実施され、製造者が市場に出回っている類似の機器に基づくすべての関連安全情報を持っていることを保証するものです。
- 財務証明書または開示書類 製造者が臨床的証拠を提出する場合は、治験責任者による開示書類を同封すること。FDAフォーム3454またはフォーム3455を参照してください。
- 適合宣言書と総括報告書。 ここでは、任意のコンセンサス規格の使用に関連する情報、またはそのような規格の一般的な使用の根拠を提供しなければならない。
- デバイスの説明 デバイスのデザイン、モデルやアクセサリーの簡単な説明はセクションに含まれるべきです。
- エグゼクティブサマリー/プレディケートの比較 このセクションでは、機器の簡単な説明、適応症や技術、機器の比較表を掲載することをお勧めします。
- 実質的な同等性の議論。 実質的な同等性を証明するために、メーカーのデバイスとプレディケート・デバイスとの詳細な比較を行います。
- 提案されたラベリング これには、21 CFR 807.87(e)に準拠した医療機器のラベリング案や、体外診断用機器の場合は21 CFR 809.10の要件に準拠したラベリング案が含まれます。
- 滅菌とシェルフライフ このセクションに記載される滅菌方法、関連するバリデーション、および主張する保存期間。
- 生体適合性。 研究プロトコル、レポート、および生体適合性研究が適正検査基準に従って実施されたことを保証する。FDAは、生体適合性研究のためのISO 10993の使用を推奨している。
- ソフトウェアです。 機器にソフトウェアが組み込まれている場合は、本項を適用する。
- 電磁両立性と電気安全性。 主に電気機器やアクティブ機器に適用される。FDAは、一般的な安全性試験にANSI/AAMI(ES)60601-1を使用するか、同等の方法を推奨しています。
- パフォーマンステスト - ベンチ メーカーや第三者機関で実施された様々な性能試験を含めることをお勧めします。これらの試験には、機械的試験、工学的試験、生物学的試験の結果が含まれますが、これらに限定されるものではありません。
- 性能試験 - 動物 動物実験が行われ、提出物に含まれている場合、FDAはその実験について説明し、性能特性を裏付ける結果を提供することを推奨しています。
- パフォーマンステスト - 臨床 提出物に臨床データ/研究が含まれている場合、FDAは、臨床研究のプロトコルと目的、試験方法、試験のエンドポイント、臨床研究で使用された統計ツールに関する情報を含めることを期待しています。
FDAの21 CFR Part 807 Subpart-Eには、製造者が事業所登録やデバイスリストを作成する際の手順が記載されていますが、510(k)申請フォーマットには、標準的なテンプレートやオールインワンの記入方法はありませんのでご注意ください。また、510(k)申請の準備に役立つ関連書式は、FDAの公式リンクからダウンロードできます。 https://www.fda.gov/medical-devices/premarket-notification-510k/510k-forms .510(k)の承認前の施設検査を行うことはありませんが、メーカーは、21 CFR Part 820の要件に従って堅牢な品質システムを導入し、万が一に備えて検査に備える必要があります。
また、特定の低~中程度のリスクの機器に対しては、「第三者審査プログラム」と呼ばれる規定があります。その名の通り、510(k)申請書を直接審査するのはFDAではなく、FDAに承認された認定された第三者機関が審査を行います。認定された第三者機関によるレビューと勧告に基づき、FDAは510(k)クリアランスの決定を下します。現在、このような第三者機関は10社あります。提出者または製造者は、自社の機器が第三者審査を受ける資格があるかどうかを以下のサイトで確認することが賢明です。 https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfpcd/classification.cfm
510k申請スケジュール
FDAに510(k)を提出する際には、510(k)のeCopyを作成し、ドキュメントコントロールセンター(DCC)のCDRHまたはCBERに提出する必要があります。提出された情報に基づいて、メーカーは異なる審査段階で追加情報の提出を求められます(図3参照)。
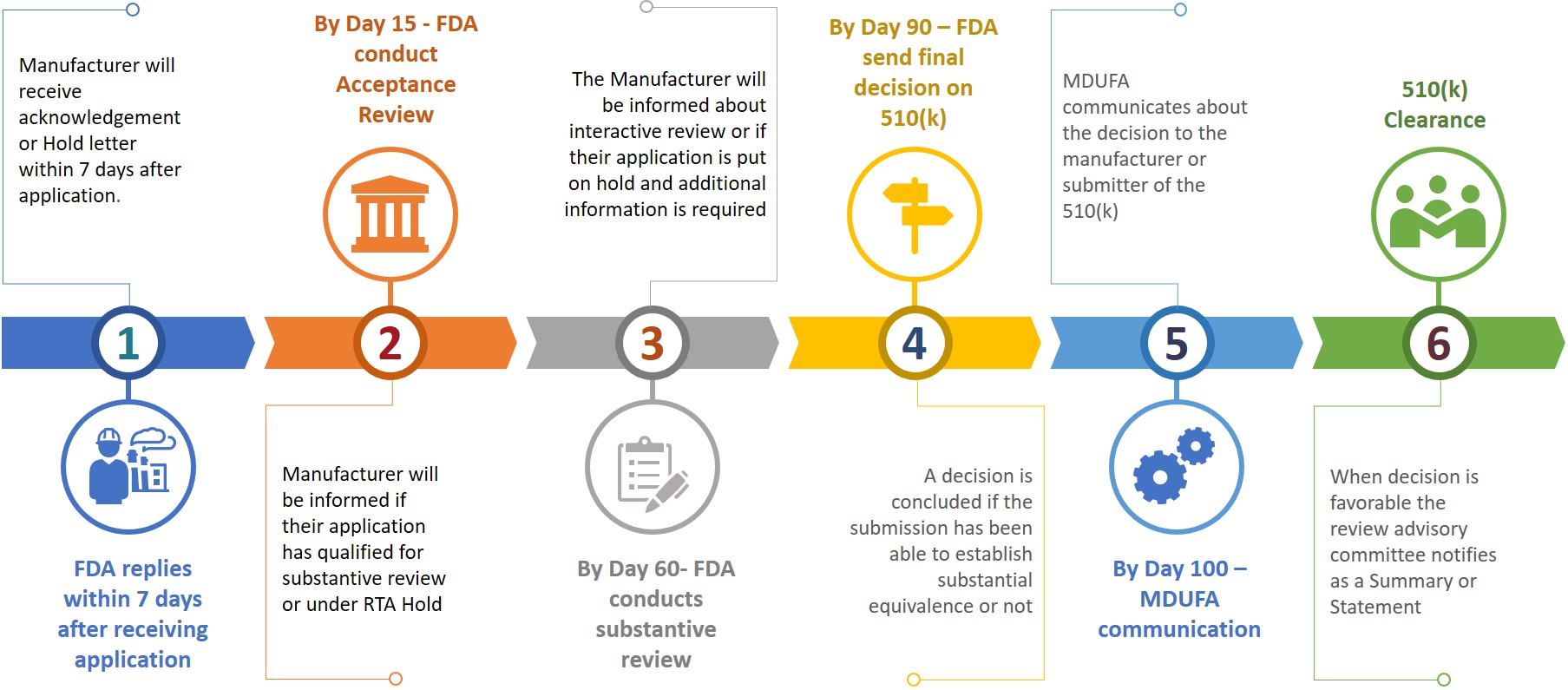
図3:レビューの暫定的なタイムライン
RTAホールドを受け取った場合、またはFDAが追加情報を求めた場合、提出者は180暦日以内に回答しなければなりません。許可された18日以内に問題が解決されない場合は、レビューシステムから自動的に削除されるか、取り下げられたとみなされます。510(k)が削除または取り下げられた場合、申請者は必要な費用を支払って新たな申請を行う必要があります。その際、同じデバイスに対する新たな申請にK番号を引用することができます。510(k)認証は、申請後100日以内に取得することができますが、認証取得に6~9ヶ月かかる場合もあります。
リファレンス
- The 510(k) Program:Evaluating Substantial Equivalence in Premarket Notifications [510(k)] Guidance for Industry and Food and Drug Administration Staff Document issued on:2014年7月28日に発行されました。
- Format for Traditional and Abbreviated 510(k)s Guidance for Industry and Food and Drug Administration Staff; Document issued on September 13, 2019.
- [1]国際貿易庁の産業・分析ユニット(I&A)と共同で作成した「医療技術のスポットライト(米国)」の概要 https://www.selectusa.gov/medical-technology-industry-united-states (最終アクセス日:2021年6月11日)

著者について
Sundeep Agarwalです。 サブジェクト・マター・エキスパートおよびFDA、CE(MDR & IVDR)コンサルタント
With a decade of experience, he is globally sought-after Leader, Speaker & Consultant in the field of QA & RA, Quality Management System, Product Design & Development, リスクマネジメント, Commercial Scale-up, Industrial Manufacturing and Clinical Studies of medical devices.An active member of a Technical Group (Software as Medical Device) at Asian Harmonization Working Party.He joins Medical Device industry/government, collaborated conferences a speaker and panelist frequently on ISO 13485, EU MDR, IVDR, CE Certification, CER, PMS, USFDA, 510(K), ISO 14971, MDSAP, Combination Devices, 人工知能 , etc. He prominently serves as a guest lecturer in various MBA and Pharmacy educational institutions in India. Kolabtreeでのプロジェクトは、彼に直接連絡してください。.
Kolabtree helps businesses worldwide hire freelance scientists and industry experts on demand. Our freelancers have helped companies publish research papers, develop products, analyze data, and more. It only takes a minute to tell us what you need done and get quotes from experts for free.

Unlock Corporate Benefits
• Secure Payment Assistance
• Onboarding Support
• Dedicated Account Manager
Sign up with your professional email to avail special advances offered against purchase orders, seamless multi-channel payments, and extended support for agreements.


