



FDA提出書類 コンサルタント、レギュレーションライター Samradni Patil を提供しています。 510k提出物チェックリスト to help 医療機器 companies with quick and easy FDA clearance.
510(k)申請プロセスは、通常、クラスIIの医療機器が米国食品医薬品局(FDA)の許可を得るために使用されます。市販承認(PMA)プロセスは、通常、クラスIIIの医療機器に使用されます。
510(k)審査では、類似の合法的に販売されているデバイス(プレディケートデバイスとも呼ばれる)との実質的同等性(SE)を判定します。実質的に同等であると主張するためには、その機器が、合法的に販売されている機器と少なくとも同等の安全性と有効性を有している必要があります。510(k)審査の対象となる機器は、プレディケート機器とのSEを主張するために、以下の点を示す必要があります。
- 合法的に販売されている機器(プレディケート・デバイス)と同じ使用目的であるプレディケート
- プレディケート・デバイスと同じ技術的特性を持つ または
- 技術的特徴や、プレディケート・デバイスと同等の安全性・有効性を示唆する情報・テストが異なること、プレディケート・デバイスとは異なる安全性・有効性に関する問題が提起されていないこと。
上記の基準を満たさない場合、Non-Substantial Equivalence(NSE)判定となります。
510k提出物チェックリスト
FDAの510(k)審査プロセスは、大きく分けて2つのステップで構成されています。
- 受入審査
- 実質的なレビュー
レビュータイムライン
| レビュータイプ | タイムライン(カレンダー日 | プロセス・アウトカム |
| 受入審査 | 15日目まで | 申請が受理されたかどうかは、FDAから申請者に通知されます。 実質的なレビュー または RTAホールドになった |
| 実質的なレビュー | 60日目まで | インタラクティブレビュー または 追加資料請求 |
注)Day1とは、FDAが510(k)申請を受理した日のことです。
まず、これらの審査の過程で、医療機器メーカーがどのような問題に直面しているのかを説明します。
受入審査
この段階で510(k)が受理されなかった場合、510(k)は RTA(Refuse to Accept)ホールド. のとおりです。 FDAデータ, 2018年には約30%の510(k)がRTAホールドされました。
実質的なレビュー
下のグラフは、実体審査の段階でFDAが出した追加情報要求の割合を示しています。
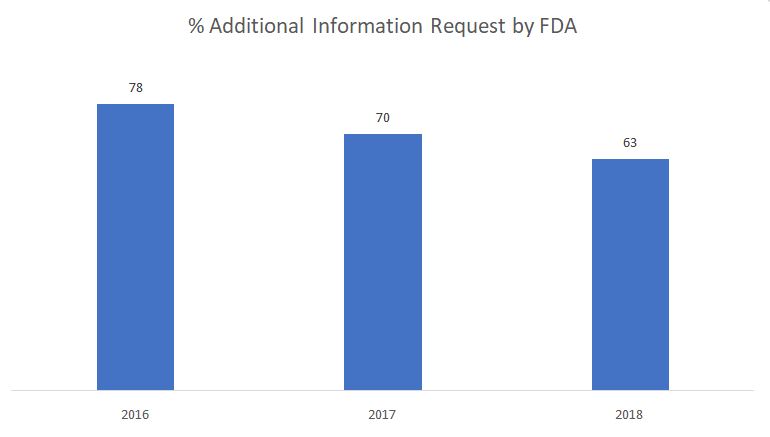
出典はこちら。 FDA
として決定した510ksの割合。 実質的に同等でないもの(NSE は以下の通りです。
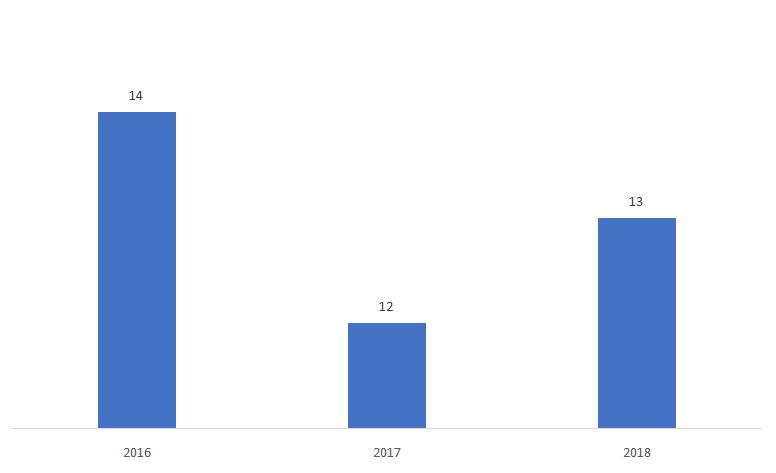
出典はこちら。 FDA
510k提出用チェックリストの共通追加情報要求
510k審査の一環として提起される一般的な問題を理解したところで、どのようなタイプの追加情報要求が多いのかに焦点を当ててみましょう。
のです。 FDAデータ は、以下のタイプの追加情報要求を描いています。
- 不適切なデバイスの説明
- 提出物全体での不一致 - このカテゴリーでの不一致の多くは、機器の説明や使用上の適応に関するものです。
- 効能・効果の問題
- 現行のガイダンス文書や認識されている基準に従わない、またはその他の方法で対処すること
- 特定のデバイスタイプに必要な性能テストがまったく行われていない(つまり、性能データがまったく提供されていない)。
- 特定のデバイスタイプに必要な臨床データが全くない(臨床データが全く提供されていない)。
ここでは、510k申請書を作成・提出する際に従うべきベストプラクティスについて説明します。
510k提出用チェックリスト
1.RTA(Refuse to Accept)レター
初期段階での受理審査の目的は、510k申請書が管理上完全であるかどうかを確認することです。ガイダンス文書 " " に記載されている受理チェックリストを確認することを強くお勧めします。510ksのポリシーを受け入れることを拒否する".
受け入れ審査を成功させるために、各企業はガイダンス文書の次の表の要素に従うことが推奨されます。
- 予備的な質問表 このチェックリストは主任審査官が最初の判断をするためのものですが、FDAに申請する前にこれらの質問に非公式に答えることを強くお勧めします。
- 組織要素表。 これらの要素は、510(k)申請書の情報を容易に識別できるように510(k)を整理するのに役立ちます。
- 完全な提出物の要素(RTA項目) 表:この表に記載されている要素は、RTAレターを取得しないための重要な要素であるため、企業はこの表に記載されている要素に細心の注意を払う必要があります。
2.不適切なデバイスの説明
510(k)申請では、デバイスの説明が必須です。このセクションには、簡単な説明と技術仕様を追加することをお勧めします。すべての医療機器のモデルおよび付属品を含める必要があります。各コンポーネントの写真、図、寸法、図面、公差を含める必要があります。重要なモデルや付属品が欠けていると、混乱を招き、追加の質問を受ける可能性があります。技術仕様が不適切な場合、誤解を招き、追加のテストを要求されることがあります。
3.提出物全体で一貫性のない情報
- デバイスの説明に矛盾がある。 モデルを追加するために510(k)申請を行うことを決定した場合、カバーレター、機器の説明、ラベリング、実質的な同等性に関する議論、性能関連セクションなどの該当セクションを実際の変更点と一致させることが重要です。一貫性がないと、管理上の遅延や、最悪の場合は追加試験の要求につながる可能性があります。
- 効能に矛盾がある。 機器の説明と同様に、510(k)の様々なセクションでの使用目的の不一致が問題となる場合があります。使用目的の記述は、SEの判断をする上で非常に重要です。
様々なセクションでの不整合は、FDAに提出する前に提出物を慎重に検討することで簡単に回避することができます。このようなミスを避けるために、様々なセクションを見るための余分な目を持つことは、常に良いアイデアです。
4.プレディケート・デバイスと異なる使用目的
FDAからSE判定を受けるためには、以下の条件を満たす必要があります。 プレディケート・デバイスと同じ使用目的.これは、プレディケートデバイスと異なる意図された使用が、異なる安全性と有効性の問題につながる可能性があるため、重要です。このような場合、510(k)はプロダクトクリアランスを得るための適切な経路ではない可能性があります。なお、プレディケートデバイスと510(k)審査中のデバイスとの間の使用目的の違いは、必ずしも使用目的の違いにはならないことに留意する必要があります。企業は、適応症の違いが使用目的の違いにつながらないことを明確に示すために、さらなる努力をする必要があります。
FDAのガイダンス "510(k)プログラム。市販前の届出における実質的同等性の評価[510(k)意図された使用関連の質問を明確にするためには、「」を参照する必要があります。
5.テスト情報の不備・欠落
- 不十分なテスト情報
特定の機器に適用される試験を理解することは重要です。機器の安全性や有効性を主張するためには、機器の種類に応じて、電気安全性試験、電磁両立性(EMC)試験、生体適合性試験、ソフトウェアバリデーション試験、滅菌試験、ユーザビリティ試験などが必要となります。
多くの場合、企業は必要なテストの量を過小評価したり、特定のテストを実施しないための理由付けをしようとします。
例。企業は、自社製品の生体適合性を主張するために、類似デバイスの生体適合性データに依存することがある。このアプローチは場合によっては受け入れられるかもしれない。しかし、多くの場合、これらのデバイス間の製造プロセスでは、510(k)審査の下でデバイスに対する個別の生体適合性試験が必要となる場合があります。
このような追加試験には数週間かかる場合があります。もしFDAが510(k)レビュー中にこれらの追加試験の実施を要求した場合、最終的な510(k)クリアランスに多くの時間を要することになります。
Appropriate teams should give thorough consideration to current FDA guidance, product design, risk management process to make determination about amount of testing required.
- テスト情報の欠落。
従来の510(k)とスペシャル510(k)の違いを理解することが重要です。 すべてのテストデータは、従来の510(k)提出物に含まれていなければなりません。
6.フォロー不足やその他の対応 現在 ガイダンス文書または公認規格
上述したように、最新の認識された規格に準拠していることを示すためのテストを確実に実施すること。最新版の規格は、旧版と比較して大幅に変更されている可能性があります。そのため、最新版の規格でテストされていない場合は、安全性と有効性について新たな疑問が生じる可能性があります。
最新のガイダンス文書を参照することで、FDAの期待値や推奨事項を理解することができます。また、提出物が査読者の理解しやすいフォーマットで書かれているため、FDAの査読プロセスが容易になります。
7.NSEの決定
510(k)プロセスの最終目標は、先行デバイスとの実質的同等性(SE)を決定することです。実体審査の段階でFDAから追加情報を要求された場合、企業は各要求を慎重に検討し、科学的に正しい回答を提供する必要があります。要求されたデータや回答を提供しなかった場合、NSE判定となる可能性があります。 多くのNSE判定は、パフォーマンスデータを提供していないことが原因です。
この段階で期待されていることを理解するために、FDAと相談し、協力することを強くお勧めします。その他、私の経験から気づいたよくある間違いをご紹介します。
管理面について
8.正しい住所での申請書の提出
これは簡単に回避できるヒューマンエラーです。必ずFDAのサイトを参照して 正しいアドレス をクリックしてご応募ください。
9.最新のFDA勧告に基づくハードコピーとeコピーを含む
FDAは、510(k)申請に必要なハードコピーとeコピーの枚数を規定しています。現在のところ、ハードコピー1部とeコピー1部が必要とされています。仮定の話をしないでください。結論を出す前に、FDAのウェブサイトを参照してください。
10. eCopyに関する問題
eCopyの技術基準を満たしている必要があります。 を参照してください。 eコピーガイダンス をクリックすると、申請書のeCopyが作成されます。
eCopy hold letterを避けるためには、PDFの命名規則とファイルサイズの推奨に従わなければなりません。の使用はできませんが eSubmitter-eCopiesツール は任意ですが、このツールはFDAの勧告に沿ってeCopyを検証するのに役立ちます。
11.審査員への情報提供
FDAから最初のホールドレター(RTAホールド、eCopyホールド)を受け取った後、企業はレビューアに情報を提出する際に間違いを犯すことがよくあります。 FDAから届いたメールやFDAの適切なガイダンスを確認して、ホールドレターに対する回答をどこに送ればよいかを確認してください。
技術的側面
12.従来の510(k)と特別な510(k)の比較
企業は、申請書を従来の510(k)または特別な510(k)に分類するという間違いを犯すことがあります。 従来の510(k)とスペシャル510(k)の大きな違いは、FDAによる申請の審査に要する時間です。スペシャル510(k)は30日、従来型510(k)は90日かかります。 私は、FDAがスペシャル510(k)を従来の510(k)に変換するよう企業に要求しているのを何度か見たことがあります。この場合、企業は変換プロセスで非常に多くの時間を費やすことになります。
FDAでは、以下のような場合にスペシャル510(k)が適切であるとしています。
- 提案された変更は、既存の機器を販売することが法的に認められているメーカーによって提出されます。
- パフォーマンスデータが不要であるか、パフォーマンスデータが必要な場合は、変化を評価するための確立された方法が利用可能であること。
- 実質的な同等性を裏付けるのに必要なすべての性能データは、要約またはリスク分析の形式でレビューすることができます。
To avoid such mistake, do thorough リサーチ on the FDA database to identify if similar change was submitted as Special 510(k) or Traditional 510(k). Refer to the ガイダンス文書 FDAから。それでも疑問がある場合は、規制当局のコンサルタントの助けを借りてください。リスクベースのアプローチを行う。それでも疑問がある場合は、保守的なアプローチをとり、従来の510(k)を提出することをお勧めします。
ユニークなチャレンジ
13.デバイスの性質
デバイスによっては、そのユニークな性質のためにユニークな問題が発生する場合があります。以下のような特定の技術分野 人工知能(AI)について そして サイバーセキュリティ は比較的新しい分野です。FDAは業界と協力してこれらの分野のガイダンス文書を作成した。
このような場合には、510(k)申請前にFDAとの事前打ち合わせを行うことを強くお勧めします。詳細は FDAガイダンス文書 510(k)申請前にFDAへのフィードバックやミーティングが必要な場合。
結論
これらは、510(k)提出物のチェックリストにおける重要なポイントでした。FDAからのRTA(Refuse to Accept)レターは、提出書類を慎重に検討し、FDAのガイダンス文書に従うことで、容易に回避することができた。
明瞭で簡潔な510(k)を書くことで、実質的な審査プロセスの一環として要求される追加情報を減らすことができます。これは、科学、芸術、経験の組み合わせであることが多いです。経験豊富な専門家の助けを借りることを強くお勧めします。 薬事コンサルタント 510(k)申請時のチェックリストに従うことで、コストのかかるミスを避けることができます。申請を成功させる可能性を高めるために、適用される規制、基準、ガイダンス文書について常に最新の情報を得るようにしてください。
に相談する必要があります。 FDA申請のエキスパート?経験豊富なレギュラトリーライター、医療機器業界の専門家と協力して 510kコンサルタント 医療機器メーカーがFDAクリアランスを成功させるための規制文書作成を支援してきました。
Kolabtree helps businesses worldwide hire freelance scientists and industry experts on demand. Our freelancers have helped companies publish research papers, develop products, analyze data, and more. It only takes a minute to tell us what you need done and get quotes from experts for free.

Unlock Corporate Benefits
• Secure Payment Assistance
• Onboarding Support
• Dedicated Account Manager
Sign up with your professional email to avail special advances offered against purchase orders, seamless multi-channel payments, and extended support for agreements.


