



Sundeep Agarwal, dispositivo medico regulatory consultant on Kolabtree, shares the essential requirements of a FDA 510k premarket notification to ensure success.
Con l'attuale $156 dimensioni del mercato da un miliardo di euro, e si prevede $208 miliardi [1] by 2023, the US medical device market is undoubtedly lucrative. Additionally, the aging population, history of chronic diseases, the assistenza sanitaria system and disruption in supply chain encourages global manufacturer to invest and expand their business horizon in the United States. Like any other global manufacturer, if you think you can leave an imprint in the world largest medical device market, you have just hit on the right blog to prepare and understand the regulatory requirement with respect to FDA’s 510(k)for your device which is utmost vital for the application process.
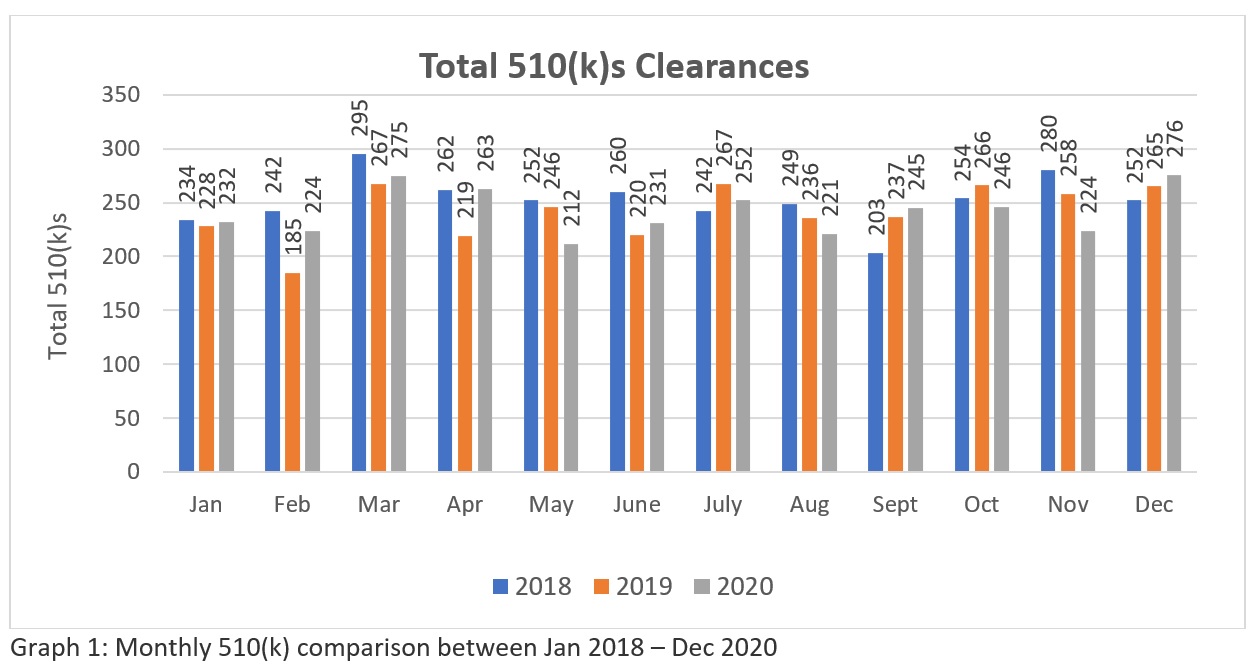
A trend analysis of three years FDA’s 510k clearances data (Source: https://www.fda.gov/medical-devices/device-approvals-denials-and-clearances/510k-clearances) rivelano che un totale di 3025-510(k) swere cleared tra gennaio 2018 a dicembre 2018, 2894 - 510(k) tra gennaio 2019 e dicembre 2019 e 2901 - 510(k) autorizzati tra gennaio 2020 e dicembre 2020. Queste cifre (vedi tabella 1) suggeriscono che attualmente, la FDA ha il potenziale per cancellare più di duecento 510(k) al mese per l'industria per soddisfare le esigenze del mercato sanitario negli Stati Uniti. Mentre la rappresentazione grafica (cfr. grafico 1) indica la coerenza della FDA nel suo approccio di revisione in mezzo alla pandemia globale. Anche se il numero non è significativamente grande rispetto al grande numero di applicazioni fatte a livello globale. Speriamo che la FDA aumenti le sue risorse in futuro. Pertanto, i produttori richiedono un'attenta preparazione e presentazione per assicurarsi di passare al primo tentativo.
| Anno 2018 | Anno 2019 | Anno 2020 | |
| Totale 510(k) | 3025 | 2894 | 2901 |
| Totale con riassunti | 2885 | 2735 | 2759 |
| Totale con dichiarazioni | 140 | 159 | 142 |
Tabella 1: La tabella indica le 510(k) annuali autorizzate tra gennaio 2018 e dicembre 2020
Requisiti FDA 510k: Trovare le chiavi
A meno che non si conosca bene la FDA Preparazione e presentazione 510(k) processo, può essere davvero stressante e impegnativo. Se un produttore di dispositivi intende commercializzare un dispositivo negli Stati Uniti che è di classe I, II e III ma non un esente da una 510(k) o che fa non richiedono una domanda di approvazione pre-mercato (PMA), allora è ammissibile per un 510(K). Procedendo oltre, il fabbricante dovrebbe fare chiarezza sui termini "dispositivo predicato" e "equivalenza sostanziale (SE)". Un dispositivo legalmente commercializzato negli Stati Uniti al quale un produttore vuole rivendicare un'equivalenza è generalmente noto come dispositivo predicato. D'altra parte, equivalenza sostanziale significa stabilire un confronto basato sull'evidenza per un nuovo dispositivo che un produttore vuole immettere nel mercato statunitense è sicuro ed efficace come il dispositivo predicato esistente, in particolare per quanto riguarda lo stesso uso previsto, le caratteristiche tecnologiche. Nel caso in cui il nuovo dispositivo differisca nelle caratteristiche tecnologiche, non dovrebbe differire in termini di sicurezza ed efficacia e il produttore è in grado di dimostrare che il dispositivo è sicuro ed efficace come il dispositivo esistente legalmente commercializzato.
Sbloccare il processo
Alcuni dei fattori fondamentali per una 510(k) di successo coinvolgono la corretta classificazione del codice prodotto, l'identificazione e la disponibilità di un dispositivo predicato nel mercato statunitense, un confronto di equivalenza sostanziale ben pianificato con prove, un robusto sistema di gestione della qualità, controlli di progettazione, una stretta aderenza agli attuali moduli FDA e il miglior utilizzo della lista di controllo Refuse to accept (RTA) (fare riferimento anche alla figura 1). La lista di controllo Refuse to accept (RTA) guida sui vari criteri di accettazione che la FDA segue mentre esegue una revisione sostanziale di una 510(k). Per tutti e tre i tipi di 510(k), ci sono diversi tipi di RTA disponibili. La lista di controllo RTA è accessibile da qui.
Il produttore dovrebbe notare che non è il numero di pagine o semplicemente la presentazione dei risultati dei test o degli studi a farli ottenere un Autorizzazione 510(k) piuttosto una scrittura ben spiegata e sequenziale (come raccomandato dalla FDA nel loro documento guida) basata sulla giustificazione scientifica e sulle prove che saranno attentamente esaminate dalla FDA entro un periodo di tempo stabilito, alla fine ottiene la 510(k). La maggior parte delle 510(k) sono respinte a causa della mancanza di chiarezza o di prove inadeguate per rivendicare un'equivalenza sostanziale [come nella 510(k) tradizionale] o un confronto insufficiente con gli standard di conformità riconosciuti dalla FDA [come nella 510(k) abbreviata]. Si osserva che l'incapacità di gestire i controlli di progettazione e la mancanza di un sistema di qualità ben stabilito è un'altra grande ragione di tale fallimento. Infine, l'indisponibilità di un team competente o la mancanza di una chiara comprensione del regolamento è un fattore innegabile che contribuisce al rifiuto oltre a quanto sopra.

Figura 1: Punti chiave da considerare per una Notifica Premarket 510(k)
Tipi di FDA 510(k) Premarket Notification
In generale, ci sono tre tipi di 510(k) che un produttore può presentare alla FDA (vedi figura 2). Essi sono:
(1) Tradizionale 510(k) - La maggior parte delle 510(k) sono in questo tipo di applicazione,
(2) Speciale 510(k) - Richiesto solo quando vengono apportate modifiche all'etichetta (o alle etichette) o al design o alcuni cambiamenti nelle indicazioni per l'uso in un dispositivo esistente precedentemente autorizzato. Il contenuto dovrebbe soddisfare i requisiti definiti in 21 CFR Parte 807.87 e 21 Parte 807.90,
(3) Abbreviato 510(k) - Si applica nel caso in cui un produttore sia in grado di produrre rapporti sull'uso di un controllo speciale o una guida o una dichiarazione di conformità basata su standard riconosciuti dalla FDA.

Figura 2: Tipi di Notifica Pre-Mercato 510(k)
Il processo di presentazione 510k
Prima di una presentazione, il produttore dovrà registrare la propria organizzazione presso la FDA. Il processo è definito come registrazione dello stabilimento secondo 21 CFR Part 807 dopo un pagamento di tasse direttamente alla FDA e lo stesso deve essere rinnovato annualmente. Per l'attuale anno finanziario 2021, la tassa è $ 5.546 per una registrazione di stabilimento. Una volta dovrebbe controllare le tasse esatte visitando il sito ufficiale della FDA user fee programs.
La FDA raccomanda 20 sezioni in una 510(k) tradizionale o abbreviata, ma non necessariamente tutte le sezioni devono essere applicabili a un produttore. A volte, se un'informazione in una sezione particolare non si applica al loro dispositivo, possono includere il titolo della sezione e scrivere "Questa sezione non si applica" o "N/A" sotto lo stesso. Le sezioni principali raccomandate del 510(k) come consigliato nel documento guida della FDA sono elencate di seguito:
- Foglio di copertura della tassa per i dispositivi medici (modulo FDA 3601): Indica una ricevuta di una tassa d'uso pagata alla FDA dal produttore.
- Foglio di copertura per la presentazione della revisione pre-mercato del Center for Devices and Radiological Health (CDRH) (modulo FDA 3514): Questo è un modulo volontario per fornire tutti i tipi di informazioni amministrative alla FDA sull'organizzazione e la presentazione.
- Una lettera di accompagnamento 510(k): Una descrizione dello scopo, del contenuto e delle informazioni amministrative sulla 510(k) dovrebbe essere incorporata in questa lettera. Si raccomanda di fare riferimento all'appendice A di "Format for Traditional and Abbreviated 510(k) s Guidance for Industry and Food and Drug Administration Staff; dtd September 13, 2019".
- Dichiarazione sulle indicazioni d'uso (modulo FDA 3881): Dovrebbe essere uniforme in tutta la 510(k). Dovrebbe anche definire se il dispositivo deve essere commercializzato come uso su prescrizione o da banco (OTC).
- Riassunto 510(k) o dichiarazione 510(k): Da preparare in conformità con il 21 CFR parte 807. Qui ci si aspetta che il produttore riassuma la 510(k) e incorpori le informazioni sul resto del contenuto.
- Dichiarazione di veridicità e accuratezza: È una dichiarazione di una persona autorizzata dell'organizzazione che certifica che tutte le informazioni presentate alla FDA relative alla 510(k) sono veritiere e accurate.
- Riassunto e certificazione di classe III: Applicabile solo ai dispositivi di classe III. È un riassunto della sicurezza e dell'efficacia e una garanzia che è stata effettuata una ricerca ragionevole e che il fabbricante ha tutte le informazioni di sicurezza pertinenti basate su dispositivi simili commercializzati.
- Certificazione finanziaria o dichiarazione di divulgazione: Se un produttore presenta prove cliniche, deve essere allegata una dichiarazione di divulgazione da parte dello sperimentatore clinico. Si può fare riferimento al modulo FDA 3454 o al modulo 3455.
- Dichiarazioni di conformità e rapporti riassuntivi: Qui vengono fornite informazioni relative all'uso di norme di consenso volontario o alla base dell'uso generale di tali norme.
- Descrizione del dispositivo: Una breve descrizione del design del dispositivo, dei modelli o degli accessori devono essere inclusi nella sezione.
- Riassunto esecutivo/confronto dei predicati: Una breve descrizione del dispositivo, le indicazioni per l'uso e la tecnologia insieme alla tabella di confronto dei dispositivi è raccomandata in questa sezione.
- Discussione sull'equivalenza sostanziale: Un confronto dettagliato tra il dispositivo del produttore e il dispositivo predicato per dimostrare l'equivalenza sostanziale.
- Etichettatura proposta: Includerà l'etichettatura proposta per il dispositivo medico secondo il 21 CFR 807.87(e) o secondo i requisiti del 21 CFR 809.10 nel caso di un dispositivo diagnostico in vitro.
- Sterilizzazione e durata di conservazione: Il metodo di sterilizzazione, la convalida pertinente e la durata di conservazione dichiarata da includere in questa sezione.
- Biocompatibilità: Protocollo degli studi, rapporti e garanzia che gli studi di biocompatibilità sono stati eseguiti seguendo le buone pratiche di laboratorio. La FDA raccomanda l'uso di ISO 10993 per gli studi di biocompatibilità.
- Software: Se il dispositivo incorpora un software, questa sezione è applicabile.
- Compatibilità elettromagnetica e sicurezza elettrica: Applicabile principalmente per dispositivi elettrici o attivi. FDA raccomanda l'uso di ANSI/AAMI (ES) 60601-1 per i test di sicurezza generale o un metodo equivalente.
- Test delle prestazioni - Banco: Si raccomanda di includere i vari test di performance condotti dal produttore o in un laboratorio terzo, che possono includere ma non limitarsi ai risultati di test meccanici o ingegneristici o biologici.
- Performance Testing - Animale: Se sono stati condotti studi sugli animali e sono inclusi nella presentazione, la FDA raccomanda di descrivere i test e fornire i risultati che supportano le caratteristiche di prestazione.
- Test delle prestazioni - Clinica: Se la presentazione includeva dati/studi clinici, la FDA si aspetta l'inclusione di informazioni sul protocollo e l'obiettivo dello studio clinico, i metodi di prova, gli endpoint dello studio e gli strumenti statistici utilizzati nello studio clinico.
Si prega di essere consapevoli del fatto che non c'è un modello standard o all-in-one pronto a riempire il formato di applicazione 510(k); anche se la FDA 21 CFR Part 807, Subpart-E descrive la procedura e guida un produttore circa la registrazione dello stabilimento e l'elenco dei dispositivi. E inoltre diversi moduli pertinenti associati a tale presentazione che sono utili per preparare possono essere scaricati dal link ufficiale della FDA dato cioè https://www.fda.gov/medical-devices/premarket-notification-510k/510k-forms . Si consiglia di fare riferimento alle varie linee guida della FDA su 510(k), disponibili in pubblico dominio al fine di soddisfare le conformità normative e documentare di conseguenza. Anche se non è una pratica per eseguire 510(k) ispezioni di impianto pre-autorizzazione, ma il produttore deve implementare un robusto sistema di qualità come da 21 CFR Parte 820 requisito ed essere pronto per l'ispezione, proprio nel caso in cui abbia luogo.
C'è anche una disposizione nota come "Third Party Review Program" per certi dispositivi a rischio basso o moderato. Come suggerisce il nome, non è la FDA che esamina direttamente la domanda 510(k), ma un'organizzazione terza accreditata approvata dalla FDA fa il lavoro. Sulla base della revisione e della raccomandazione fatta dalla terza parte accreditata, la FDA conclude una decisione su una autorizzazione 510(k). Attualmente ci sono 10 organizzazioni terze di questo tipo disponibili. Per un presentatore o un produttore, sarà saggio controllare se il loro dispositivo è ammissibile per una revisione da parte di terzi su https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfpcd/classification.cfm
Timeline dell'applicazione 510k
Un presentatore o produttore mentre presenta un 510(k) alla FDA richiede anche di preparare una copia elettronica del loro 510(k) e presentare lo stesso al CDRH o CBER al centro di controllo dei documenti (DCC). Sulla base delle informazioni presentate, al produttore verranno chieste ulteriori informazioni durante le diverse fasi di revisione (vedi figura 3).
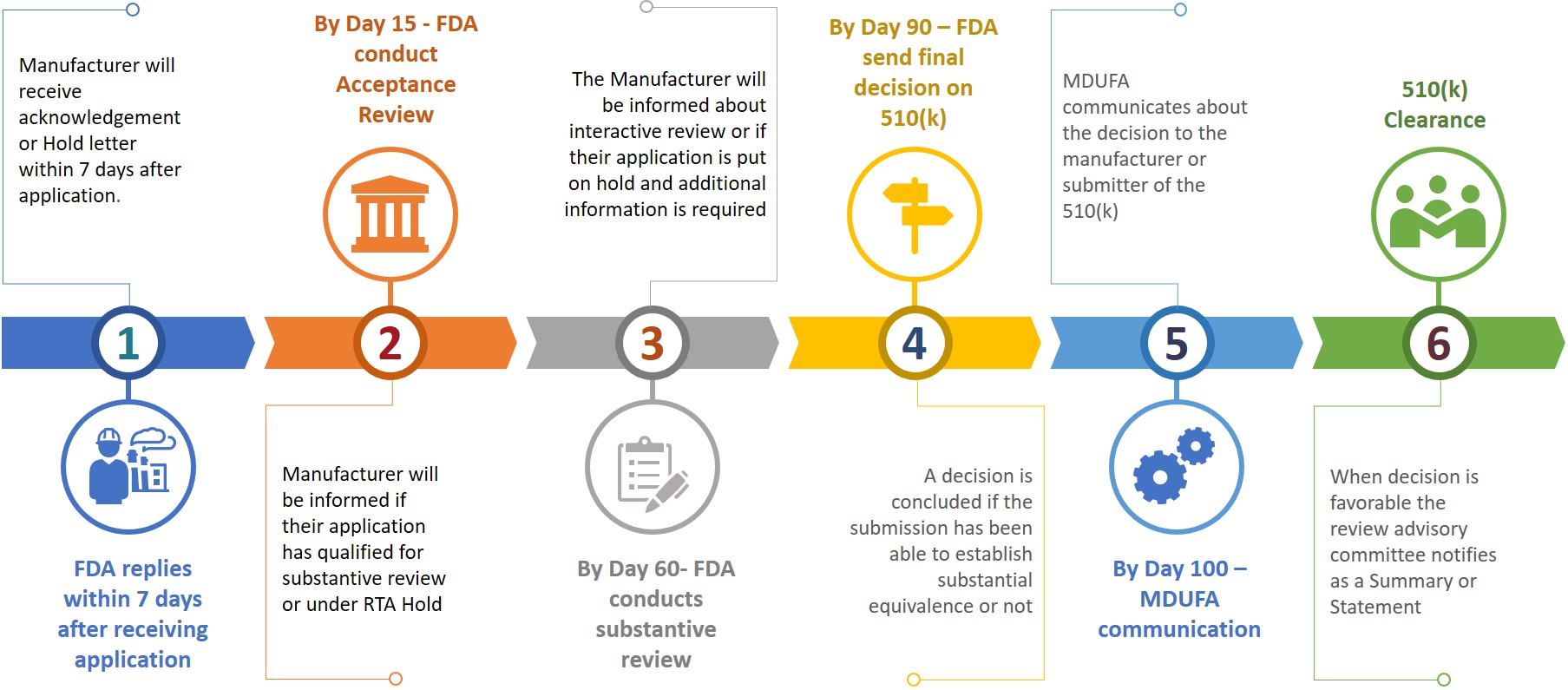
Figura 3: Tempistica provvisoria di revisione
Il presentatore avrà 180 giorni di calendario per rispondere quando riceve una sospensione RTA o se la FDA vuole ulteriori informazioni. La mancata risoluzione di qualsiasi problema entro i 18 giorni di calendario consentiti comporterà la cancellazione automatica dal sistema di revisione o sarà considerata ritirata. In caso di cancellazione o ritiro di una 510(k), il presentatore dovrà ripresentare una nuova domanda dopo aver pagato la tassa necessaria mentre il numero K può essere citato nella nuova domanda per lo stesso dispositivo. Un'autorizzazione 510(k) può essere ottenuta entro 100 giorni dalla presentazione, mentre possono essere necessari anche 6 - 9 mesi per ottenere l'autorizzazione.
Riferimenti
- Il programma 510(k): Evaluating Substantial Equivalence in Premarket Notifications [510(k)] Guidance for Industry and Food and Drug Administration Staff Document issued on: 28 luglio 2014.
- Formato per le 510(k) tradizionali e abbreviate Guida per l'industria e il personale della Food and Drug Administration; Documento pubblicato il 13 settembre 2019.
- [1]La panoramica della tecnologia medica (USA), preparata in collaborazione con l'Unità di Industria e Analisi (I&A) dell'International Trade Administration https://www.selectusa.gov/medical-technology-industry-united-states (Ultimo accesso l'11 giugno 2021)

L'autore
Sundeep Agarwal, Esperto in materia e consulente FDA, CE (MDR & IVDR)
With a decade of experience, he is globally sought-after Leader, Speaker & Consultant in the field of QA & RA, Quality Management System, Product Design & Development, Gestione del rischio, Commercial Scale-up, Industrial Manufacturing and Clinical Studies of medical devices.An active member of a Technical Group (Software as Medical Device) at Asian Harmonization Working Party.He joins Medical Device industry/government, collaborated conferences a speaker and panelist frequently on ISO 13485, EU MDR, IVDR, CE Certification, CER, PMS, USFDA, 510(K), ISO 14971, MDSAP, Combination Devices, Intelligenza artificiale , etc. He prominently serves as a guest lecturer in various MBA and Pharmacy educational institutions in India. Contattatelo direttamente per un progetto su Kolabtree.
Kolabtree helps businesses worldwide hire freelance scientists and industry experts on demand. Our freelancers have helped companies publish research papers, develop products, analyze data, and more. It only takes a minute to tell us what you need done and get quotes from experts for free.

Unlock Corporate Benefits
• Secure Payment Assistance
• Onboarding Support
• Dedicated Account Manager
Sign up with your professional email to avail special advances offered against purchase orders, seamless multi-channel payments, and extended support for agreements.


