



A rapport d'évaluation clinique (CER) is a technical document required by dispositif médical companies that want to sell or distribute products in Europe. The CER provides a comprehensive overview of the dispositif médical design and composition, intended usage and applications, les essais cliniques l'analyse et les résultats, les analyses documentaires pertinentes, les protocoles et les instructions d'utilisation.
MDR de l'UE et marquage CE
Medical devices in Europe need to get a “CE Mark”, which indicates that the product complies with the EU regulations (MDD, MDR or IVDR, whichever is applicable). The recent Medical Device Regulations in Europe has significantly impacted the way medical devices are regulated and approved in the EU. The MDR is the biggest change to medical device legislation in over 20 years. Some major changes in the MDR (as compared with the MDD) include device classification, technical file documentation, traceability and post-market surveillance.
Lire la suite : Conseils pour obtenir un marquage CE pour votre dispositif médical
Rapport d'évaluation clinique
The MDR places great emphasis on Clinical Evaluation Reports or CERs. The CER forms part of the Technical File, which is the main documentation which demonstrates conformité réglementaire and provides all the information about the device. Medical device companies need to take several measures to make sure that their CER is well-prepared and well-maintained. CER documentation typically comprises 4 stages:
- Définir le champ d'application du dispositif, son utilisation prévue et les allégations thérapeutiques/diagnostiques.
- Identifier et valider les données cliniques
- Analyser les données et les interpréter pour voir si elles répondent à toutes les exigences.
- Identifier les risques et les incertitudes, auxquels il est possible de répondre au cours de la surveillance post-commercialisation (PMS).
Une CER doit être régulièrement mise à jour tout au long du cycle de vie d'un dispositif médical. Le document doit contenir les preuves cliniques qui étayent la preuve de la conformité aux Exigences Essentielles (ERs) du MEDDEV 2.7/1 Rev. 4 Annexe 1 (Exigences de sécurité et de performance dans le MDR). Le document doit également décrire les aspects physiques et techniques et la composition du dispositif, ainsi que les instructions d'utilisation.
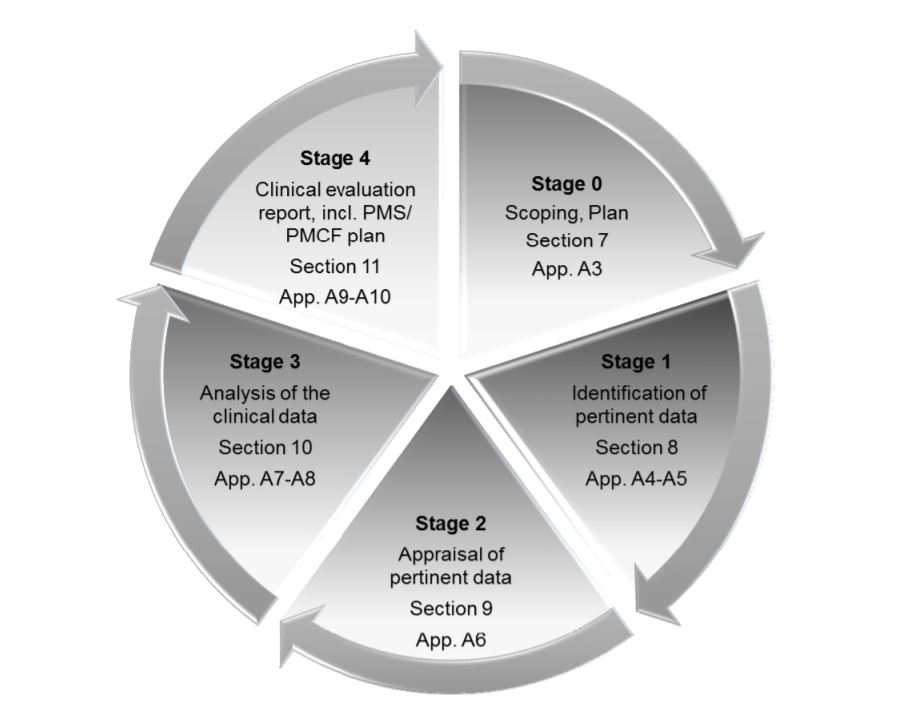
Les quatre étapes de la RCE, telles que décrites dans MEDDEV 2.7/ rev. 4.
Équivalence
Les fabricants de dispositifs médicaux doivent évaluer les données cliniques de leur propre dispositif médical ou d'un dispositif équivalent, dont la sécurité et les performances cliniques ne sont pas différentes. En vertu du RIM, le fabricant de dispositifs médicaux doit tenir compte de trois facteurs pour prouver qu'un produit est équivalent : biologique, technique et clinique.
Équivalence technique signifie que le dispositif doit avoir les mêmes spécifications de conception et de composition, et doit être utilisé dans les mêmes conditions, avec la même méthode de déploiement et les mêmes principes opérationnels. Équivalence clinique signifie que le dispositif doit être utilisé pour traiter la même affection, au même endroit du corps et pour le même groupe de population, en obtenant des performances similaires.
Équivalence biologique signifie que le dispositif médical en question ne doit pas entraîner de risque biologique.
Si un dispositif médical n'est pas en mesure de démontrer l'équivalence, il doit soit effectuer des recherches cliniques supplémentaires, soit retirer l'allégation de son produit.
Revue de la littérature
Les résultats de la recherche documentaire constituent une partie essentielle du rapport d'évaluation clinique. Un processus solide d'examen de la documentation doit être mis en place afin que toute la documentation publiée relative au dispositif soit facilement accessible et puisse soutenir l'allégation faite. Les revues de la littérature nécessitent généralement l'expertise d'un chercheur qualifié qui peut identifier le terme de recherche correct, utiliser des bases de données comme PubMED ou MEDLine, trouver la littérature pertinente et l'évaluer, y compris en utilisant les références pertinentes. L'analyse documentaire doit également aborder toutes les questions auxquelles les données cliniques ne peuvent répondre.
Combien de temps faut-il pour rédiger un CER ?
L'identification et la collecte de toutes les données qui entrent dans la CER et la préparation du document conformément aux directives peuvent prendre plusieurs semaines ou mois. La surveillance post-commercialisation (PMS) constitue une part importante du processus, même après la certification du marquage CE, et la PMS doit être un élément central du système de gestion de la qualité (QMS) du fabricant. Dans certains cas, un suivi clinique post-commercialisation (PMCF) est également requis, ce qui implique la collecte proactive de données cliniques et leur évaluation pour répondre aux exigences de sécurité et de performance.
La CER n'est pas un document statique. Il est recommandé aux fabricants de dispositifs médicaux d'élaborer un processus et une stratégie solides intégrant les bonnes pratiques pour les aider à conserver l'accès au marché de l'UE, à éviter tout rappel de produits et à rationaliser le temps et les ressources consacrés à la tenue du document.
Confier la rédaction de CER à un expert indépendant peut vous faire gagner un temps et une énergie précieux. Rédacteurs indépendants de rapports d'évaluation clinique sont en mesure de développer des CER à partir de 3 000 USD sur Kolabtree.
Rédacteurs de rapports d'évaluation clinique
The time and expertise required to put together a CER can be a resource strain for several medical device startups or medical device SMBs, who may not have in-house specialists. Working with external experts, including freelance CER writers, clinical data analysts, literature review experts biostatisticians and les rédacteurs médicaux can help companies make sure that their CER strategy is robust and their data is all verified and accurate.
Comment engager un rédacteur CER indépendant
Les compétences à rechercher chez un rédacteur de CER comprennent une expérience des réglementations et des processus d'approbation des dispositifs médicaux, une bonne connaissance de MEDDEV 2.7/ rev. 4, la connaissance des domaines thérapeutiques et de solides compétences analytiques. L'équipe mondiale d'experts de Kolabtree comprend des personnes qualifiées dans les domaines suivants Rédacteurs de rapports d'évaluation cliniquequi peuvent vous aider à établir un rapport d'évaluation exhaustif et bien documenté. Il est gratuit de publier votre projet et d'obtenir des devis d'experts. Commencez
Experts connexes :
Consultants en dispositifs médicaux | Rédacteur médical indépendant | Rédacteur indépendant du CER | Consultants MDR de l'UE | Soumissions à la FDA | Experts en recherche documentaire | Experts en conformité réglementaire | Expert en essais cliniques | Rédacteurs de textes réglementaires | Consultants PMS | Consultants du PMCF | Experts en biostatistique | Expert en développement de produits
Kolabtree helps businesses worldwide hire freelance scientists and industry experts on demand. Our freelancers have helped companies publish research papers, develop products, analyze data, and more. It only takes a minute to tell us what you need done and get quotes from experts for free.

Unlock Corporate Benefits
• Secure Payment Assistance
• Onboarding Support
• Dedicated Account Manager
Sign up with your professional email to avail special advances offered against purchase orders, seamless multi-channel payments, and extended support for agreements.


