



Sundeep Agarwal, dispositif médical regulatory consultant on Kolabtree, shares the essential requirements of a FDA 510k premarket notification to ensure success.
Avec l'actuel $156 milliards d'euros de taille de marché, et prévu $208 milliards d'euros [1] by 2023, the US medical device market is undoubtedly lucrative. Additionally, the aging population, history of chronic diseases, the soins de santé system and disruption in supply chain encourages global manufacturer to invest and expand their business horizon in the United States. Like any other global manufacturer, if you think you can leave an imprint in the world largest medical device market, you have just hit on the right blog to prepare and understand the regulatory requirement with respect to FDA’s 510(k)for your device which is utmost vital for the application process.
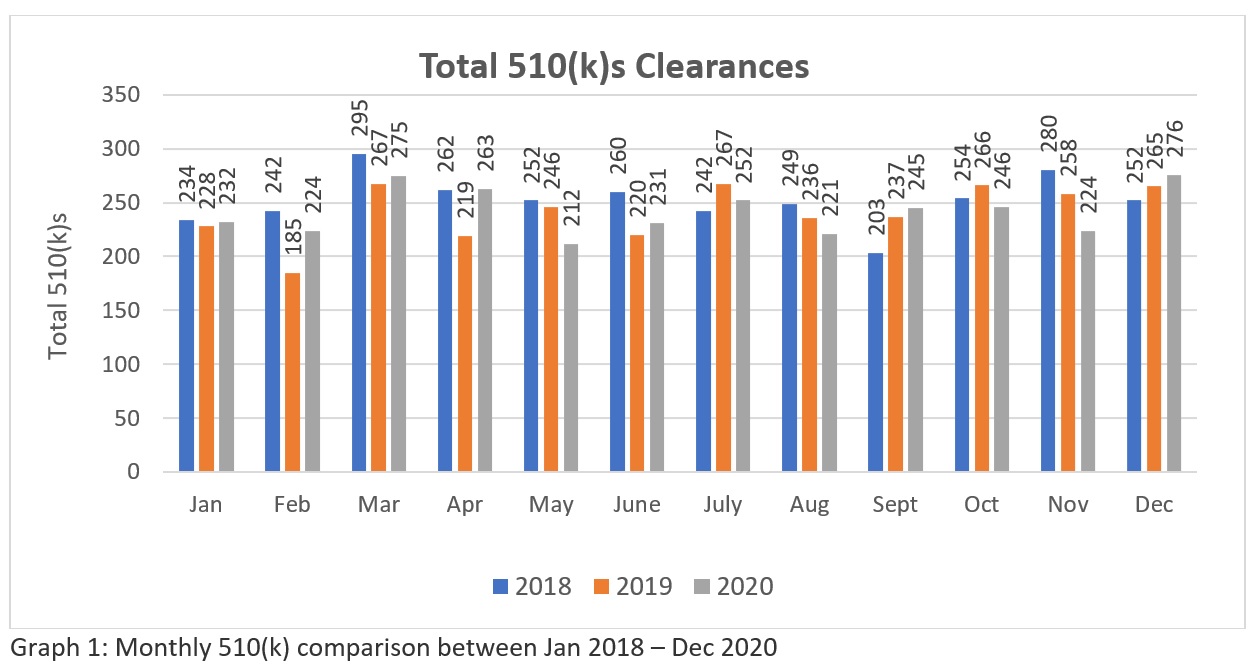
A trend analysis of three years FDA’s 510k clearances data (Source: https://www.fda.gov/medical-devices/device-approvals-denials-and-clearances/510k-clearances) révèlent qu'un total de 3025-510(k)swere dégagé entre janvier 2018 et décembre 2018, 2894 - 510(k)s entre janvier 2019 et décembre 2019 et 2901 - 510(k)s approuvés entre janvier 2020 et décembre 2020. Ces chiffres (voir tableau 1) suggèrent qu'actuellement, la FDA a le potentiel d'autoriser plus de deux cents 510(k) par mois pour permettre à l'industrie de répondre aux besoins du marché de la santé aux États-Unis. La représentation graphique (voir graphique 1) montre la cohérence de l'approche de la FDA en matière d'examen dans le contexte de la pandémie mondiale. Bien que le nombre ne soit pas très élevé par rapport au grand nombre de demandes faites dans le monde. Nous espérons que la FDA augmentera ses ressources à l'avenir. Par conséquent, les fabricants doivent se préparer soigneusement et soumettre leurs demandes afin de s'assurer qu'elles soient acceptées dès la première tentative.
| Année 2018 | Année 2019 | Année 2020 | |
| Total des 510(k)s | 3025 | 2894 | 2901 |
| Total avec résumés | 2885 | 2735 | 2759 |
| Total avec déclarations | 140 | 159 | 142 |
Tableau 1 : Le tableau indique les autorisations annuelles 510(k) obtenues entre janvier 2018 et décembre 2020.
Exigences 510k de la FDA : Trouver les clés
A moins de bien connaître les règles de la FDA Préparation et soumission de la demande 510(k) le processus peut être vraiment stressant et difficile. Si un fabricant de dispositifs a l'intention de commercialiser aux États-Unis un dispositif de classe I, II ou III, mais pas de classe I, II ou III, il doit obtenir une autorisation de mise sur le marché. une exonération d'un 510(k) ou qui ne ne pas exiger une demande d'autorisation de mise sur le marché (PMA), il est alors éligible pour un 510(K). Pour aller plus loin, le fabricant doit avoir une idée claire des termes "dispositif prédicat" et "équivalence substantielle (SE)". Un dispositif légalement commercialisé aux États-Unis et pour lequel un fabricant souhaite revendiquer une équivalence est généralement connu sous le nom de dispositif prédicat. D'autre part, l'équivalence substantielle signifie l'établissement d'une comparaison fondée sur des preuves pour un nouveau dispositif qu'un fabricant veut mettre sur le marché américain est sûr et efficace comme le dispositif prédicat existant est particulièrement en ce qui concerne la même utilisation prévue, les caractéristiques technologiques. Si le nouveau dispositif diffère par ses caractéristiques technologiques, il ne doit pas différer en termes de sécurité et d'efficacité et le fabricant est en mesure de démontrer que le dispositif est aussi sûr et efficace que le dispositif existant légalement commercialisé.
Déverrouiller le processus
Parmi les facteurs essentiels à la réussite d'un 510(k) figurent une classification correcte du code produit, l'identification et la disponibilité d'un dispositif prédicat sur le marché américain, une comparaison d'équivalence substantielle bien planifiée avec des preuves, un système de gestion de la qualité robuste, des contrôles de conception, une adhésion stricte aux formulaires actuels de la FDA et la meilleure utilisation de la liste de contrôle de refus d'acceptation (RTA) (voir également la figure 1). La liste de contrôle de refus d'acceptation (RTA) donne des indications sur les différents critères d'acceptation que la FDA suit lorsqu'elle effectue un examen de fond d'un 510(k). Pour les trois types de 510(k), il existe différents types de RTA. La liste de contrôle RTA est accessible à partir de ici.
Le fabricant doit noter que ce n'est pas le nombre de pages ou le simple fait de soumettre les résultats de tests ou d'études qui lui permettraient d'obtenir un prix. Autorisation 510(k) Le 510(k) est plutôt le résultat d'une rédaction séquentielle bien expliquée (comme le recommande la FDA dans son document d'orientation), basée sur des justifications et des preuves scientifiques qui seront soigneusement examinées par la FDA dans un délai déterminé. La majorité des 510(k) sont rejetés en raison d'un manque de clarté ou de preuves insuffisantes pour revendiquer une équivalence substantielle [comme dans le cas du 510(k) traditionnel] ou d'une comparaison insuffisante avec les normes de conformité reconnues par la FDA [comme dans le cas du 510(k) abrégé]. On observe que l'incapacité à gérer les contrôles de conception et l'absence d'un système de qualité bien établi constituent une autre raison importante de cet échec. Enfin, l'indisponibilité d'une équipe compétente ou le manque de compréhension de la réglementation est un facteur indéniable qui contribue au rejet en plus de ce qui précède.

Figure 1 : Points clés à considérer pour une notification préalable à la mise sur le marché 510(k)
Types de notification de précommercialisation 510(k) de la FDA
En général, il existe trois types de 510(k)s qu'un fabricant peut soumettre à la FDA (voir figure 2). Il s'agit de :
(1) Traditionnel 510(k) - La plupart des 510(k) sont dans ce type de demande,
(2) Spécial 510(k) - Nécessaire uniquement lorsque des modifications sont apportées à l'étiquette ou à la conception ou certaines modifications de l'indication d'utilisation d'un dispositif existant précédemment autorisé. Le contenu doit répondre aux exigences définies dans 21 CFR Part 807.87 et 21 Part 807.90,
(3) Abrégé 510(k) - S'applique dans le cas où un fabricant est en mesure de produire des rapports sur l'utilisation d'un contrôle ou d'un guide spécial ou une déclaration de conformité basée sur les normes reconnues de la FDA.

Figure 2 : Types de notification préalable à la mise sur le marché 510(k)
Le processus de soumission 510k
Avant de soumettre un dossier, le fabricant devra enregistrer son organisation auprès de la FDA. Le processus est appelé enregistrement d'établissement conformément à 21 CFR Part 807, après paiement des frais directement à la FDA, qui doivent être renouvelés chaque année. Pour l'exercice financier 2021, les frais sont de $ 5,546 pour l'enregistrement d'un établissement. Vous pouvez vérifier le montant exact des frais en consultant le site officiel des programmes de frais d'utilisation de la FDA.
La FDA recommande 20 sections dans un 510(k) traditionnel ou abrégé, mais toutes les sections ne sont pas nécessairement applicables à un fabricant. Parfois, si une information dans une section particulière ne s'applique pas à leur dispositif, ils peuvent inclure le titre de la section et écrire "Cette section ne s'applique pas" ou "N/A" sous le même titre. Les principales sections recommandées du formulaire 510(k), telles que conseillées dans le document d'orientation de la FDA, sont énumérées ci-dessous :
- Feuille de couverture du Medical Device User Fee (formulaire FDA 3601) : Indique la réception d'un droit d'utilisation payé à la FDA par le fabricant.
- Feuille de couverture de la demande d'examen de précommercialisation du Center for Devices and Radiological Health (CDRH) (formulaire FDA 3514) : Il s'agit d'un formulaire volontaire permettant de fournir à la FDA toutes sortes d'informations administratives sur l'organisation et la soumission.
- Une lettre de présentation 510(k) : Une description de l'objectif, du contenu et des informations administratives concernant le 510(k) doit être incorporée dans cette lettre. Il est recommandé de se référer à l'annexe A du document "Format for Traditional and Abbreviated 510(k)s Guidance for Industry and Food and Drug Administration Staff ; dtd September 13, 2019".
- Déclaration d'indication d'utilisation (formulaire FDA 3881) : Elle doit être uniforme dans l'ensemble du 510(k). Elle doit également définir si le dispositif doit être commercialisé sur ordonnance ou en vente libre.
- Résumé 510(k) ou Déclaration 510(k) : A préparer conformément à 21 CFR part 807. Ici, le fabricant est censé résumer le 510(k) et incorporer des informations sur le reste du contenu.
- Déclaration de véracité et d'exactitude : Il s'agit d'une déclaration d'une personne autorisée de l'organisation certifiant que toutes les informations soumises à la FDA concernant le 510(k) sont véridiques et exactes.
- Résumé et certification de la classe III : Applicable uniquement aux dispositifs de classe III. Il s'agit d'un résumé de la sécurité et de l'efficacité et de l'assurance qu'une recherche raisonnable a été effectuée et que le fabricant dispose de toutes les informations de sécurité pertinentes basées sur des dispositifs similaires commercialisés.
- Certification financière ou déclaration de divulgation : Si un fabricant soumet des preuves cliniques, une déclaration de divulgation par l'investigateur clinique doit être jointe. Le formulaire 3454 ou 3455 de la FDA peut être utilisé.
- Déclarations de conformité et rapports de synthèse : Les informations relatives à l'utilisation des normes de consensus volontaire ou à la base de l'utilisation générale de ces normes doivent être fournies ici.
- Description du dispositif : Une brève description de la conception de l'appareil, des modèles ou des accessoires doit être incluse dans la section.
- Résumé/Comparaison des prédicats : Une brève description du dispositif, des indications d'utilisation et de la technologie ainsi qu'un tableau comparatif des dispositifs sont recommandés dans cette section.
- Discussion sur l'équivalence substantielle : Une comparaison détaillée entre le dispositif du fabricant et le dispositif prédicat pour démontrer l'équivalence substantielle.
- Étiquetage proposé : Il comprendra l'étiquetage proposé pour le dispositif médical conformément à la norme 21 CFR 807.87(e) ou aux exigences de la norme 21 CFR 809.10 dans le cas d'un dispositif de diagnostic in vitro.
- Stérilisation et durée de conservation : La méthode de stérilisation, la validation pertinente et la durée de conservation revendiquée doivent être incluses dans cette section.
- Biocompatibilité : Protocole d'études, rapports et assurance que les études de biocompatibilité ont été réalisées conformément aux bonnes pratiques de laboratoire. La FDA recommande l'utilisation de la norme ISO 10993 pour les études de biocompatibilité.
- Logiciel : Si le dispositif intègre un logiciel, la présente section est applicable.
- Compatibilité électromagnétique et sécurité électrique : Applicable principalement aux dispositifs électriques ou actifs. La FDA recommande l'utilisation de la norme ANSI/AAMI (ES) 60601-1 pour les tests de sécurité générale ou une méthode équivalente.
- Test de performance - Banc : Il est recommandé d'inclure les différents tests de performance effectués par le fabricant ou dans un laboratoire tiers, qui peuvent inclure, sans s'y limiter, les résultats de tests mécaniques, d'ingénierie ou biologiques.
- Test de performance - Animal : Si des études sur les animaux ont été menées et sont incluses dans la soumission, la FDA recommande de décrire les tests et de fournir les résultats qui soutiennent les caractéristiques de performance.
- Test de performance - clinique : Si la demande comprend des données/études cliniques, la FDA s'attend à ce que soient incluses des informations sur le protocole et l'objectif de l'étude clinique, les méthodes d'essai, les paramètres de l'étude et les outils statistiques utilisés dans l'étude clinique.
Sachez qu'il n'existe pas de modèle standard ou de format tout-en-un prêt à être rempli pour la demande 510(k). Cependant, le document 21 CFR Part 807, Subpart-E de la FDA décrit la procédure et guide le fabricant pour l'enregistrement de l'établissement et l'inscription du dispositif. En outre, plusieurs formulaires pertinents associés à une telle soumission et utiles à la préparation peuvent être téléchargés à partir du lien officiel de la FDA, à savoir https://www.fda.gov/medical-devices/premarket-notification-510k/510k-forms . Bien qu'il ne soit pas d'usage d'effectuer des inspections 510(k) préalables à l'autorisation, le fabricant doit mettre en œuvre un système de qualité robuste conformément aux exigences de la norme 21 CFR Part 820 et être prêt à l'inspection, au cas où elle aurait lieu.
Il existe également une disposition connue sous le nom de "Programme d'examen par un tiers" pour certains dispositifs à risque faible ou modéré. Comme son nom l'indique, ce n'est pas la FDA qui examine directement la demande 510(k), mais une organisation tierce accréditée et approuvée par la FDA qui fait ce travail. Sur la base de l'examen et des recommandations de la tierce partie accréditée, la FDA prend une décision concernant l'autorisation 510(k). Il existe actuellement 10 organisations tierces de ce type. Pour un soumissionnaire ou un fabricant, il sera judicieux de vérifier si son dispositif peut faire l'objet d'un examen par une tierce partie à l'adresse suivante https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfpcd/classification.cfm
Calendrier de la demande 510k
Un soumissionnaire ou un fabricant qui soumet un 510(k) à la FDA doit également préparer une copie électronique de son 510(k) et la soumettre au CDRH ou au CBER au centre de contrôle des documents (DCC). Sur la base des informations soumises, le fabricant devra fournir des informations supplémentaires au cours des différentes étapes de révision (voir figure 3).
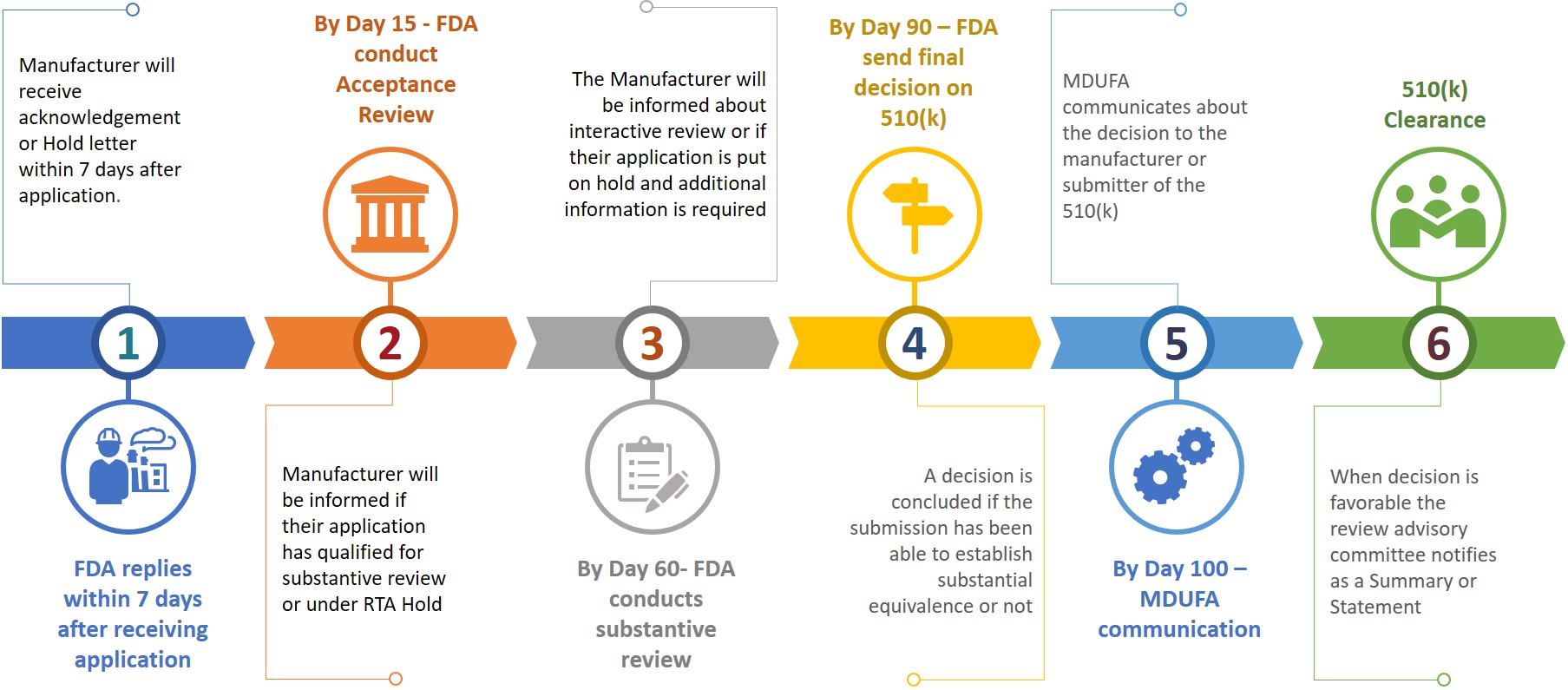
Figure 3 : Calendrier provisoire de l'examen
Le soumissionnaire disposera de 180 jours civils pour répondre lorsqu'il reçoit une mise en attente RTA ou si la FDA souhaite des informations supplémentaires. Si les problèmes ne sont pas résolus dans les 18 jours civils autorisés, le système de révision sera automatiquement supprimé ou considéré comme retiré. En cas de suppression ou de retrait d'un 510(k), le soumissionnaire devra refaire une nouvelle demande après avoir payé les frais nécessaires, le numéro K pouvant être cité dans la nouvelle demande pour le même dispositif. Une autorisation 510(k) peut être obtenue dans les 100 jours suivant la soumission de la demande, mais il faut parfois attendre 6 à 9 mois pour l'obtenir.
Références
- Le programme 510(k) : Evaluating Substantial Equivalence in Premarket Notifications [510(k)] Guidance for Industry and Food and Drug Administration Staff Document publié le : 28 juillet 2014.
- Format pour les 510(k)s traditionnels et abrégés Guide pour l'industrie et le personnel de la Food and Drug Administration ; Document publié le 13 septembre 2019.
- [1]L'aperçu de la technologie médicale (États-Unis), préparé en collaboration avec l'unité Industrie et analyse (I&A) de l'administration du commerce international. https://www.selectusa.gov/medical-technology-industry-united-states (Dernière consultation le 11 juin 2021)

A propos de l'auteur
Sundeep Agarwal, Expert en la matière et consultant FDA, CE (MDR et IVDR)
With a decade of experience, he is globally sought-after Leader, Speaker & Consultant in the field of QA & RA, Quality Management System, Product Design & Development, Gestion des risques, Commercial Scale-up, Industrial Manufacturing and Clinical Studies of medical devices.An active member of a Technical Group (Software as Medical Device) at Asian Harmonization Working Party.He joins Medical Device industry/government, collaborated conferences a speaker and panelist frequently on ISO 13485, EU MDR, IVDR, CE Certification, CER, PMS, USFDA, 510(K), ISO 14971, MDSAP, Combination Devices, Intelligence artificielle , etc. He prominently serves as a guest lecturer in various MBA and Pharmacy educational institutions in India. Contactez-le directement pour un projet sur Kolabtree.
Kolabtree helps businesses worldwide hire freelance scientists and industry experts on demand. Our freelancers have helped companies publish research papers, develop products, analyze data, and more. It only takes a minute to tell us what you need done and get quotes from experts for free.

Unlock Corporate Benefits
• Secure Payment Assistance
• Onboarding Support
• Dedicated Account Manager
Sign up with your professional email to avail special advances offered against purchase orders, seamless multi-channel payments, and extended support for agreements.


