



Soumissions à la FDA consultant et rédacteur réglementaire Samradni Patil fournit un Liste de contrôle pour les soumissions 510k to help dispositif médical companies with quick and easy FDA clearance.
Le processus de soumission 510(k) est généralement utilisé pour les dispositifs médicaux de classe II afin d'obtenir l'autorisation de la Food and Drug Administration (FDA) américaine. Le processus d'approbation préalable à la mise sur le marché (PMA) est généralement utilisé pour les dispositifs médicaux de classe III.
Le processus d'examen 510(k) détermine l'équivalence substantielle (ES) avec un dispositif similaire légalement commercialisé, également appelé dispositif prédicat. Le dispositif doit être au moins aussi sûr et efficace que le dispositif légalement commercialisé pour pouvoir prétendre qu'il est substantiellement équivalent à ce dernier. Le dispositif soumis à l'examen 510(k) doit présenter les éléments suivants pour prétendre à l'équivalence avec le dispositif prédicat :
- Même utilisation prévue que le dispositif légalement commercialisé (dispositif prédicat)prédicat
- Les mêmes caractéristiques technologiques que le dispositif prédicat ou
- Des caractéristiques technologiques différentes et des informations/essais suggérant que le dispositif est aussi sûr et efficace que le dispositif prédicat et des questions différentes sur la sécurité et l'efficacité que le dispositif prédicat ne sont pas soulevées.
Le non-respect des critères ci-dessus entraîne la détermination de l'équivalence non substantielle (NSE).
Liste de contrôle des soumissions 510k
Le processus d'examen 510(k) de la FDA peut être divisé en deux étapes.
- Examen de l'acceptation
- Examen de fond
Délais de révision
| Type de révision | Délai (jours calendaires) | Résultat du processus |
| Examen de l'acceptation | Au 15e jour | La FDA informe le demandeur de l'acceptation ou non de la demande pour les raisons suivantes Examen de fond ou Placé en attente de RTA |
| Examen de fond | Au jour 60 | Examen interactif ou Demande d'informations supplémentaires |
Note : Le jour 1 est le jour où la FDA reçoit la demande 510(k).
Voyons d'abord quels sont les problèmes auxquels sont confrontées les entreprises de dispositifs médicaux au cours de ces processus d'examen.
Examen de l'acceptation
Si la demande 510(k) n'est pas acceptée à ce stade, elle est placée sur le site Internet de la Commission européenne. Refus d'acceptation (RTA). Conformément à Données de la FDAEn 2018, environ 30% 510(k)s ont été placés en attente de RTA.
Examen de fond
Le graphique ci-dessous montre le pourcentage de demandes d'informations complémentaires émises par la FDA au cours de la phase de révision substantielle.
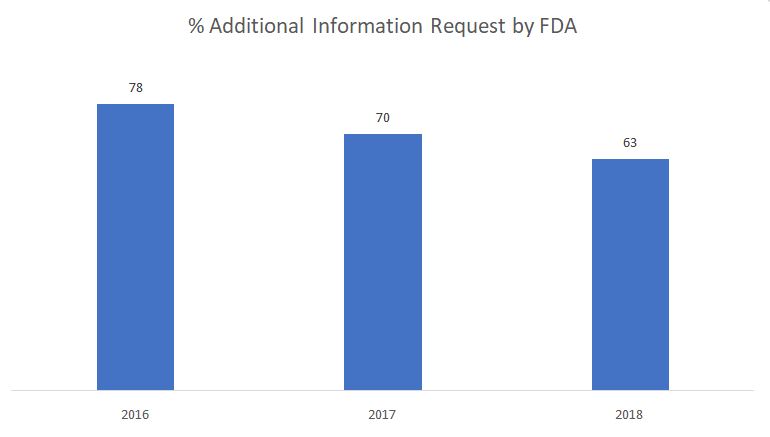
Source : FDA
Le pourcentage de 510ks déterminé comme Non-Substantiellement Équivalent (NSE) est présenté ci-dessous.
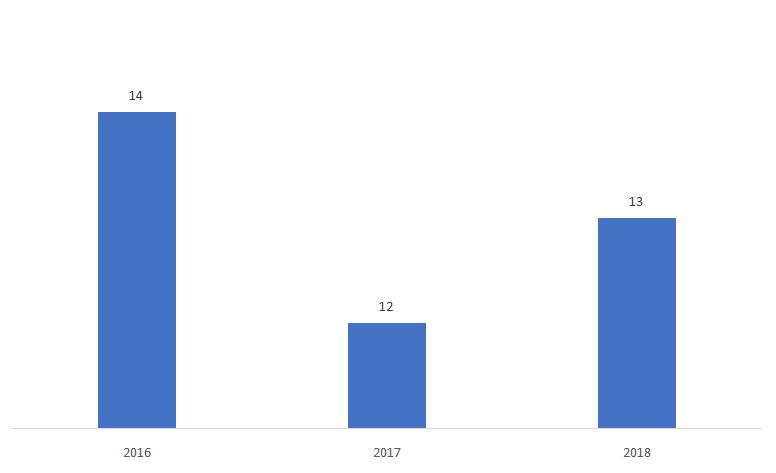
Source : FDA
Liste de contrôle des demandes d'informations supplémentaires courantes de la soumission 510k
Maintenant que nous comprenons les problèmes courants soulevés dans le cadre du processus d'examen 510k, concentrons-nous sur le type de demandes d'informations supplémentaires qui sont courantes.
Le site Données de la FDA représente les types suivants de demandes d'informations supplémentaires :
- Description inadéquate du dispositif
- Divergences dans la soumission - Les divergences dans cette catégorie sont le plus souvent liées à la description du dispositif ou aux indications d'utilisation.
- Problèmes liés aux indications d'utilisation
- Non-respect ou non-respect des documents d'orientation ou des normes reconnues en vigueur.
- Les tests de performance requis pour certains types de dispositifs sont totalement absents (c'est-à-dire qu'aucune donnée de performance n'est fournie).
- Les données cliniques requises pour certains types de dispositifs sont totalement absentes (c'est-à-dire qu'aucune donnée clinique n'est fournie).
Voyons maintenant les meilleures pratiques à suivre pour préparer et soumettre une demande 510k.
Liste de contrôle pour la soumission 510k :
1. Lettre de refus d'acceptation (RTA)
L'objectif de l'examen d'acceptation au stade initial est de vérifier si la demande 510k est administrativement complète. Il est fortement recommandé d'examiner la liste de contrôle d'acceptation fournie dans le document d'orientation "Politique de refus d'acceptation pour 510ks".
Afin d'assurer le succès de l'examen d'acceptation, il est suggéré à chaque entreprise de suivre les éléments du tableau suivant du document d'orientation.
- Tableau des questions préliminaires : Bien que cette liste de contrôle soit destinée à l'examinateur principal pour qu'il prenne une décision initiale, il est fortement recommandé de répondre à ces questions de manière informelle avant de soumettre la demande à la FDA.
- Tableau des éléments organisationnels : Ces éléments permettent d'organiser la demande 510(k) de manière à faciliter l'identification des informations dans la demande 510(k).
- Tableau des éléments d'une soumission complète (éléments du CRT): Les entreprises devraient prêter une plus grande attention aux éléments énumérés dans ce tableau, car ces éléments sont essentiels pour ne pas obtenir une lettre de RTA.
2. Description inadéquate du dispositif
La description du dispositif est obligatoire dans la demande 510(k). Il est suggéré d'ajouter une brève description et des spécifications techniques dans cette section. Tous les modèles et accessoires de dispositifs médicaux doivent être inclus. Des photos, des diagrammes, des dimensions, des dessins et des tolérances pour chaque composant doivent être inclus. L'absence d'un modèle ou d'un accessoire important peut entraîner une confusion et des questions supplémentaires. Des spécifications techniques inappropriées peuvent conduire à des idées fausses et à une demande de tests supplémentaires.
3. Informations incohérentes dans l'ensemble de la soumission
- Incohérence dans la description du dispositif : Si une entreprise décide d'effectuer une soumission 510(k) pour ajouter un modèle supplémentaire, il est important que les sections applicables telles que la lettre d'accompagnement, la description du dispositif, l'étiquetage, la discussion sur l'équivalence substantielle, les sections relatives aux performances soient alignées sur les changements réels. Une incohérence peut entraîner des retards administratifs et, dans le pire des cas, une demande de tests supplémentaires.
- Incohérence dans l'indication d'utilisation : Comme pour la description du dispositif, l'incohérence de l'indication d'utilisation dans les différentes sections du 510(k) peut poser problème. La déclaration d'indication d'utilisation est très importante pour déterminer la SE.
Les incohérences dans les différentes sections peuvent facilement être évitées par un examen minutieux de la demande avant qu'elle ne soit soumise à la FDA. Il est toujours bon d'avoir une paire d'yeux supplémentaire pour jeter un coup d'œil aux différentes sections afin d'éviter de telles erreurs.
4. Utilisation prévue différente de celle du dispositif prédicat
Pour obtenir la détermination SE de la FDA, le dispositif doit présenter les caractéristiques suivantes la même utilisation prévue que le dispositif prédicat. Ceci est important car une utilisation différente de celle du dispositif prédicat peut entraîner des problèmes de sécurité et d'efficacité différents. Dans ce cas, la procédure 510(k) peut ne pas être une voie appropriée pour obtenir l'autorisation du produit. Il convient de noter que les différences dans l'indication d'utilisation entre le dispositif prédicat et le dispositif soumis à l'examen 510(k) n'entraînent pas nécessairement une utilisation prévue différente. Les entreprises doivent faire des efforts supplémentaires pour montrer clairement que les différences dans l'indication d'utilisation n'ont pas entraîné une utilisation différente.
Le guide de la FDA "Le programme 510(k) : Évaluation de l'équivalence substantielle dans les notifications préalables à la mise sur le marché [510(k)]."Il convient de se référer à ce document pour obtenir des éclaircissements sur les questions liées à l'utilisation prévue.
5. Informations inadéquates/manquantes sur les tests
- Informations inadéquates sur les tests :
Il est important de comprendre les tests applicables à un dispositif particulier. En fonction du type de dispositif, des tests de sécurité électrique, de compatibilité électromagnétique (CEM), de biocompatibilité, de validation de logiciels, de stérilisation et d'utilisabilité peuvent être nécessaires pour démontrer la sécurité et l'efficacité d'un dispositif.
Souvent, les entreprises sous-estiment la quantité de tests nécessaires ou tentent de justifier le fait de ne pas effectuer certains tests.
Exemple : Les entreprises peuvent s'appuyer sur les données de biocompatibilité d'un dispositif similaire pour revendiquer la biocompatibilité de leur produit. Cette approche peut être acceptable dans certains cas. Cependant, il arrive souvent que le processus de fabrication entre ces dispositifs justifie des tests de biocompatibilité distincts sur le dispositif dans le cadre de l'examen 510(k).
Ces tests supplémentaires peuvent prendre plusieurs semaines. Si la FDA demande de réaliser ces tests supplémentaires pendant l'examen du dossier 510(k), cela ajoute beaucoup de temps à l'autorisation finale du dossier 510(k).
Appropriate teams should give thorough consideration to current FDA guidance, product design, risk management process to make determination about amount of testing required.
- Informations manquantes sur les tests :
Il est important de comprendre la différence entre le 510(k) traditionnel et le 510(k) spécial. Toutes les données de test doivent être incluses dans la soumission traditionnelle 510(k).
6. Défaut de suivre ou de traiter autrement Actuel Document(s) d'orientation ou normes reconnues
Comme indiqué ci-dessus, assurez-vous que les tests sont effectués pour démontrer la conformité aux dernières normes reconnues. Il peut y avoir des changements importants dans la dernière version de la norme par rapport à la version précédente. Cela peut entraîner des questions supplémentaires sur la sécurité et l'efficacité si le dispositif n'est pas testé avec la dernière version de la norme.
La consultation des derniers documents d'orientation permet de comprendre les attentes ou les recommandations de la FDA. Cela facilite également le processus d'examen de la FDA car la soumission est rédigée dans un format facilement compréhensible par l'examinateur.
7. Détermination de l'ESN
L'objectif ultime du processus 510(k) est de déterminer l'équivalence substantielle (SE) avec un dispositif prédicat. Lorsque des informations supplémentaires sont demandées par la FDA au cours de la phase de révision substantielle, les entreprises doivent examiner attentivement chaque demande et fournir une réponse scientifiquement fondée. Le fait de ne pas fournir les données/réponses demandées peut entraîner la détermination de l'ES. Beaucoup de déterminations de l'ESN sont dues à l'absence de données sur les performances.
Il est fortement recommandé de consulter et de collaborer avec la FDA pour comprendre les attentes à cette phase. Voici d'autres erreurs courantes que j'ai pu constater au cours de mon expérience.
Aspects administratifs
8. Soumission de la demande à l'adresse correcte
Il s'agit d'une erreur humaine facilement évitable. Consultez toujours le site web de la FDA pour connaître les l'adresse correcte pour soumettre votre candidature.
9. Inclure une copie papier ainsi qu'une copie électronique conformément aux dernières recommandations de la FDA.
La FDA a des exigences spécifiques concernant le nombre de copies papier et de copies électroniques qui doivent être soumises pour la demande 510(k). Actuellement, 1 copie papier et 1 copie électronique sont requises. Ne faites pas d'hypothèses. Consultez le site Web de la FDA avant de tirer des conclusions hâtives.
10. Problèmes liés à eCopy
La soumission doit répondre aux normes techniques d'eCopy. Reportez-vous à la Conseils sur la copie électronique pour préparer la copie électronique de la demande.
La convention de dénomination des PDF et la recommandation sur la taille des fichiers doivent être respectées pour éviter la lettre d'attente d'eCopy. Bien que l'utilisation du Outil eSubmitter-eCopies est volontaire, cet outil permet de valider l'eCopy conformément aux recommandations de la FDA.
11. Soumettre des informations à l'examinateur
Après avoir reçu la lettre de mise en suspens initiale de la FDA (mise en suspens RTA, mise en suspens eCopy), les entreprises commettent souvent une erreur en soumettant les informations à l'examinateur. Vérifiez le courriel reçu de la FDA ou les directives appropriées de la FDA concernant l'endroit où envoyer la réponse à la lettre de mise en suspens.
Aspects techniques
12. 510(k) traditionnel et 510(k) spécial
Les entreprises peuvent commettre l'erreur de classer la demande dans la catégorie 510(k) traditionnelle ou 510(k) spéciale. La principale différence entre le 510(k) traditionnel et le 510(k) spécial est le temps nécessaire à l'examen de la demande par la FDA. La procédure spéciale 510(k) prend 30 jours civils, tandis que la procédure traditionnelle 510(k) prend 90 jours civils. J'ai vu plusieurs fois la FDA demander aux entreprises de convertir un 510(k) spécial en 510(k) traditionnel. Dans ce cas, les entreprises finissent par perdre beaucoup de temps dans le processus de conversion.
Selon la FDA, un 510(k) spécial peut être approprié lorsque :
- La modification proposée est soumise par le fabricant légalement autorisé à commercialiser le dispositif existant ;
- Les données sur le rendement sont inutiles, ou si des données sur le rendement sont nécessaires, des méthodes bien établies sont disponibles pour évaluer le changement ; et
- Toutes les données de performance nécessaires pour étayer l'équivalence en substance peuvent être examinées sous forme de résumé ou d'analyse des risques.
To avoid such mistake, do thorough recherche on the FDA database to identify if similar change was submitted as Special 510(k) or Traditional 510(k). Refer to the documents d'orientation de la FDA. Si vous avez encore des doutes, demandez l'aide de consultants en réglementation. Utilisez une approche basée sur le risque. Si le doute persiste, il est recommandé d'adopter une approche conservatrice et de soumettre un 510(k) traditionnel.
Des défis uniques
13. Nature de l'appareil
Certains dispositifs peuvent soulever des questions uniques en raison de leur nature particulière. Certains domaines technologiques tels que Intelligence artificielle (IA) et Cybersécurité sont relativement nouvelles. La FDA a collaboré avec l'industrie pour élaborer des documents d'orientation dans ces domaines.
Dans ce cas, une réunion préalable avec la FDA avant la soumission du 510(k) est fortement recommandée. Se référer à la Document d'orientation de la FDA si les entreprises ont besoin d'un retour d'information et d'une réunion avec la FDA avant la soumission du 510(k).
Conclusion
Ce sont les points importants de la liste de contrôle d'une soumission 510(k). Les lettres de refus d'acceptation (RTA) de la FDA pouvaient facilement être évitées en examinant soigneusement la soumission et en suivant les documents d'orientation de la FDA.
La demande d'informations supplémentaires dans le cadre du processus d'examen de fond pourrait être réduite par la rédaction de 510(k) clairs et concis. Il s'agit souvent d'une combinaison de science, d'art et d'expérience. Il est fortement recommandé de recourir à l'aide de personnes expérimentées. Consultants en affaires réglementaires dans la rédaction du 510(k) pour éviter des erreurs coûteuses, suivez ces listes de contrôle pour la soumission du 510(k). Restez à jour en termes de réglementations, normes et documents d'orientation applicables pour augmenter les chances de réussite de la soumission.
Besoin de consulter un Expert en soumissions à la FDA? Travailler avec des rédacteurs expérimentés en matière de réglementation, des experts du secteur des dispositifs médicaux et des représentants de l'industrie. Consultants 510k qui ont aidé des entreprises de dispositifs médicaux à préparer des documents réglementaires pour obtenir l'autorisation de la FDA.
Kolabtree helps businesses worldwide hire freelance scientists and industry experts on demand. Our freelancers have helped companies publish research papers, develop products, analyze data, and more. It only takes a minute to tell us what you need done and get quotes from experts for free.

Unlock Corporate Benefits
• Secure Payment Assistance
• Onboarding Support
• Dedicated Account Manager
Sign up with your professional email to avail special advances offered against purchase orders, seamless multi-channel payments, and extended support for agreements.


