



Le Dr. Javier Quilez Oliete, un spécialiste expérimenté de la freelance bioinformatics consultant on Kolabtree, provides a comprehensive guide to DNA sequencing analyse des données, including tools and software used to read data.
Introduction
L'acide désoxyribonucléique (ADN) est la molécule qui porte la plupart des informations génétiques. d'un organisme. (Dans certains types de virus, l'information génétique est portée par l'acide ribonucléique (ARN)). Les nucléotides (conventionnellement représentés par les lettres A, C, G ou T) sont les unités de base des molécules d'ADN. Conceptuellement, Séquençage de l'ADN est le processus de lecture des nucléotides qui composent une molécule d'ADN (par exemple, "GCAAACCAAT" est une chaîne d'ADN de 10 nucléotides). Les technologies de séquençage actuelles produisent des millions de lectures d'ADN. dans un délai raisonnable et à un coût relativement faible. À titre de référence, le coût du séquençage d'un génome humain - un génome est l'ensemble des molécules d'ADN d'un organisme - a chuté de 1,5 million d'euros à 1,5 million d'euros. Barrière $100 et cela peut être fait en quelques jours. Cela contraste avec la première initiative de séquençage de la le génome humainqui a été achevé en une décennie et dont le coût s'élève à environ $2,7 milliards.
This capability to sequence DNA at high throughput and low cost has enabled the development of a growing number of sequencing-based methods and applications. For example, sequencing entire genomes or their protein-coding regions (two approaches known respectively as whole genome and exome sequencing) in disease and healthy individuals can hint to disease-causing DNA alterations. Also, the sequencing of the RNA that is transcribed from DNA—a technique known as RNA-sequencing—is used to quantify gene activity and how this changes in different conditions (e.g. untreated versus treatment). On the other side, chromosome conformation capture sequencing methods detect interactions between nearby DNA molecules and thus help to determine the spatial distribution of chromosomes within the cell.
Ces applications et d'autres du séquençage de l'ADN ont en commun la génération d'ensembles de données de l'ordre du gigaoctet et comprenant des millions de séquences lues. Par conséquent, pour donner un sens aux expériences de séquençage à haut débit (HTS), il faut disposer d'importantes capacités d'analyse des données. Heureusement, des outils informatiques et statistiques spécialisés et des flux d'analyse relativement standard existent pour la plupart des types de données HTS. Si certaines des étapes d'analyse (initiales) sont communes à la plupart des types de données de séquençage, l'analyse en aval dépendra du type de données et/ou de l'objectif final de l'analyse. Je vous propose ci-dessous une introduction aux étapes fondamentales de l'analyse des données HTS et je vous renvoie à des outils populaires.
Certaines des sections ci-dessous sont axées sur l'analyse des données générées par les technologies de séquençage à lecture courte (principalement les suivantes Illumina), car elles ont historiquement dominé le marché des HTS. Cependant, les nouvelles technologies qui génèrent des lectures plus longues (par ex. Oxford Nanopore Technologies, PacBio) gagnent rapidement du terrain. Comme le séquençage à long terme présente certaines particularités (par exemple, des taux d'erreur plus élevés), des outils spécifiques sont développés pour l'analyse de ce type de données.
Contrôle de qualité (CQ) des données brutes
L'analyste avide commencera l'analyse à partir des fichiers FASTQ ; le Format FASTQ est depuis longtemps la norme pour stocker les données de séquençage à lecture courte. Essentiellement, les fichiers FASTQ contiennent la séquence de nucléotides et les données par base. qualité d'appel pour des millions de lectures. Bien que la taille du fichier dépende du nombre réel de lectures, les fichiers FASTQ sont généralement volumineux (de l'ordre de mégaoctets et gigaoctets) et compressés. Il est à noter que la plupart des outils qui utilisent les fichiers FASTQ en entrée peuvent les traiter en format compressé. Ainsi, afin d'économiser de l'espace disque, il est recommandé de ne pas les décompresser. Par convention, j'assimilerai ici un fichier FASTQ à un échantillon de séquençage.
FastQC est probablement l'outil le plus populaire pour effectuer le CQ des lectures brutes. Il peut être exécuté via une interface visuelle ou par programme. Si la première option est plus pratique pour les utilisateurs qui ne se sentent pas à l'aise avec l'environnement en ligne de commande, la seconde offre une évolutivité et une reproductibilité incomparables (pensez à la pénibilité et au risque d'erreur que représente l'exécution manuelle de l'outil pour des dizaines de fichiers). Quoi qu'il en soit, le résultat principal de FastQC est un fichier Fichier HTML reporting key summary statistiques about the overall quality of the raw sequencing reads from a given sample. Inspecting tens of FastQC reports one by one is tedious and it complicates the comparison across samples. Therefore, you may want to use MultiQCqui regroupe les rapports HTML de FastQC (ainsi que d'autres outils utilisés en aval, par exemple l'ajustement des adaptateurs, l'alignement) en un seul rapport..
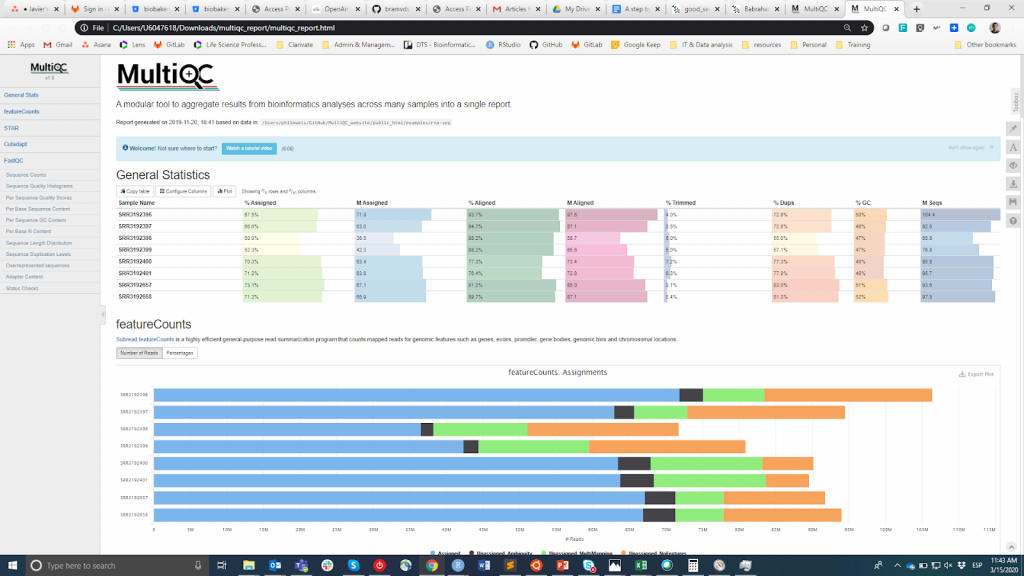
MultiQC
Les informations CQ sont destinées à permettre à l'utilisateur de juger si les échantillons sont de bonne qualité et peuvent donc être utilisés pour les étapes suivantes ou s'ils doivent être rejetés. Malheureusement, il n'existe pas de seuil consensuel basé sur les métriques FastQC pour classer les échantillons comme étant de bonne ou de mauvaise qualité. L'approche que j'utilise est la suivante. Je m'attends à ce que tous les échantillons qui sont passés par la même procédure (par exemple, l'extraction d'ADN, la préparation de la bibliothèque) aient des statistiques de qualité similaires et une majorité de drapeaux "pass". Si certains échantillons ont une qualité inférieure à la moyenne, je les utiliserai quand même dans l'analyse en aval en gardant cela à l'esprit. D'un autre côté, si tous les échantillons de l'expérience obtiennent systématiquement des drapeaux "warning" ou "fail" dans plusieurs métriques (cf. cet exemple), je soupçonne que quelque chose s'est mal passé dans l'expérience (par exemple, une mauvaise qualité d'ADN, la préparation de la bibliothèque, etc.) et je recommande de la répéter.
Lire l'habillage
Le CQ des lectures brutes permet d'identifier les échantillons problématiques, mais il n'améliore pas la qualité réelle des lectures. Pour ce faire, nous devons découper les lectures afin de supprimer les séquences techniques et les extrémités de mauvaise qualité.
Les séquences techniques sont des restes de la procédure expérimentale (par exemple, les adaptateurs de séquençage). Si ces séquences sont adjacentes à la véritable séquence de la lecture, l'alignement (voir ci-dessous) peut faire correspondre les lectures à une mauvaise position dans le génome ou diminuer la confiance dans un alignement donné. Outre les séquences techniques, on peut également vouloir éliminer les séquences d'origine biologique si elles sont très présentes parmi les lectures. Par exemple, des procédures de préparation de l'ADN non optimales peuvent laisser une forte proportion d'ARN ribosomal (ARNr) converti en ADN dans l'échantillon. À moins que ce type d'acide nucléique ne soit la cible de l'expérience de séquençage, le fait de conserver les lectures dérivées de l'ARNr ne fera qu'augmenter la charge de calcul des étapes en aval et risque de brouiller les résultats. Il convient de noter que si les niveaux de séquences techniques, d'ARNr ou d'autres contaminants sont très élevés, ce qui aura probablement déjà été mis en évidence par le CQ, il est préférable de rejeter l'ensemble de l'échantillon de séquençage.
Dans le séquençage à lecture courte, la séquence d'ADN est déterminée un nucléotide à la fois (techniquement, un nucléotide à chaque cycle de séquençage). En d'autres termes, le nombre de cycles de séquençage détermine la longueur de lecture. Un problème connu des méthodes de séquençage HTS est la diminution de la précision avec laquelle les nucléotides sont déterminés à mesure que les cycles de séquençage s'accumulent. Cela se traduit par une diminution globale de la qualité d'appel par base, en particulier vers la fin de la lecture. Comme cela se produit avec les séquences techniques, essayer d'aligner des lectures qui contiennent des extrémités de mauvaise qualité peut conduire à un mauvais placement ou à une mauvaise qualité de cartographie.
Pour supprimer les séquences techniques/contaminantes et les extrémités de mauvaise qualité, des outils d'élagage de lecture tels que Trimmomatic et Cutadapt existent et sont largement utilisés. Essentiellement, ces outils éliminent les séquences techniques (disponibles en interne et/ou fournies par l'utilisateur) et découpent les lectures en fonction de leur qualité tout en maximisant leur longueur. Les lectures qui restent trop courtes après le découpage sont écartées (les lectures excessivement courtes, par exemple <36 nucléotides, compliquent l'étape d'alignement car elles sont susceptibles de correspondre à plusieurs sites dans le génome). Vous pouvez regarder le pourcentage de lectures qui survivent à l'élagage, car un taux élevé de lectures rejetées est probablement un signe de mauvaise qualité des données.
Enfin, j'exécute à nouveau FastQC sur les lectures découpées pour vérifier que cette étape a été efficace et a systématiquement amélioré les mesures de contrôle.
Alignement
À quelques exceptions près (par exemple assemblage de novo), l'alignement (également appelé cartographie) est généralement l'étape suivante pour la plupart des types de données et des applications HTS. L'alignement des lectures consiste à déterminer la position dans le génome d'où provient la séquence de la lecture (typiquement exprimée comme chromosome:début-fin). Par conséquent, à cette étape, il est nécessaire d'utiliser une séquence de référence pour aligner/mapper les lectures.
Le choix de la séquence de référence sera déterminé par de multiples facteurs. Tout d'abord, l'espèce dont provient l'ADN séquencé. Si le nombre d'espèces pour lesquelles une séquence de référence de haute qualité est disponible augmente, ce n'est pas toujours le cas pour certains organismes moins étudiés. Dans ces cas, vous pouvez aligner les lectures sur une espèce proche sur le plan évolutif pour laquelle un génome de référence est disponible. Par exemple, comme il n'existe pas de séquence de référence pour le génome du coyote, nous pouvons utiliser celle du chien, espèce étroitement apparentée, pour l'alignement des lectures. De même, nous pouvons toujours vouloir aligner nos lectures sur une espèce étroitement apparentée pour laquelle il existe une séquence de référence de meilleure qualité. Par exemple, alors que le génome du gibbon a été publié surLe génome humain, quant à lui, est fragmenté en milliers de fragments qui ne reflètent pas entièrement l'organisation de ce génome en dizaines de chromosomes ; dans ce cas, il peut être utile d'effectuer l'alignement en utilisant la séquence de référence humaine.
Un autre facteur à prendre en compte est la version de l'assemblage de la séquence de référence, puisque de nouvelles versions sont publiées au fur et à mesure que la séquence est mise à jour et améliorée. Il est important de noter que les coordonnées d'un alignement donné peuvent varier d'une version à l'autre. Par exemple, de multiples versions du génome humain peuvent être trouvées dans la base de données de l'OMS. Navigateur génomique de l'UCSC. Dans tous les cas, je recommande fortement de migrer vers la dernière version de l'assemblage dès qu'elle est disponible. Cela peut causer quelques désagréments pendant la transition, car les résultats déjà existants seront relatifs aux anciennes versions, mais c'est payant à long terme.
En outre, le type de données de séquençage a également son importance. Les lectures générées par les protocoles DNA-seq, ChIP-seq ou Hi-C seront alignées sur la séquence de référence du génome. D'autre part, comme l'ARN transcrit à partir de l'ADN est ensuite transformé en ARNm (c'est-à-dire que les introns sont supprimés), de nombreuses lectures d'ARN-seq ne pourront pas être alignées sur une séquence de référence du génome. Au lieu de cela, nous devons soit les aligner sur des séquences de référence du transcriptome, soit utiliser des aligneurs sensibles au fractionnement (voir ci-dessous) lorsque nous utilisons la séquence du génome comme référence. Le choix de la source pour l'annotation de la séquence de référence, c'est-à-dire la base de données contenant les coordonnées des gènes, des transcrits, des centromères, etc. est lié à cette question. J'utilise généralement la base de données Annotation GENCODE car il combine une annotation complète des gènes et des séquences de transcription.
Une longue liste d'outils d'alignement de séquences à lecture courte a été développée (voir la section sur l'alignement de séquences à lecture courte). ici). Reviewing them is beyond the scope of this article (details about the algorithms behind these tools can be found ici). D'après mon expérience, les plus populaires sont les suivants Bowtie2, BWA, HISAT2, Minimap2, STAR et TopHat. Je vous recommande de choisir votre aligneur en tenant compte de facteurs clés tels que le type de données HTS. et l'application ainsi que l'acceptation par la communauté, la qualité de la documentation et le nombre d'utilisateurs. Par exemple, on a besoin d'aligneurs comme STAR ou Bowtie2 qui sont conscients des jonctions exon-exon lors du mappage de l'ARN séquentiel au génome.
La plupart des mappeurs ont en commun la nécessité d'indexer la séquence utilisée comme référence avant que l'alignement proprement dit n'ait lieu. Cette étape peut prendre du temps, mais elle ne doit être effectuée qu'une seule fois pour chaque séquence de référence. La plupart des mappeurs stockent les alignements dans des fichiers SAM/BAM, qui suivent le principe de l'indexation. Format SAM/BAM (Les fichiers BAM sont des versions binaires des fichiers SAM). L'alignement est l'une des étapes les plus longues et les plus complexes de l'analyse des données de séquençage et les fichiers SAM/BAM sont lourds (de l'ordre de plusieurs gigaoctets). Il est donc important de s'assurer que vous disposez des ressources nécessaires (voir la dernière section ci-dessous) pour exécuter l'alignement dans un temps raisonnable et stocker les résultats. De même, en raison de la taille et du format binaire des fichiers BAM, évitez de les ouvrir avec des éditeurs de texte ; utilisez plutôt des commandes Unix ou des outils dédiés tels que SAMtools.
Des alignements
Je dirais qu'il n'y a pas d'étape commune claire après l'alignement, car c'est à ce stade que chaque type de données HTS et chaque application peuvent différer.
Une analyse en aval courante pour les données ADN-seq est l'appel de variants, c'est-à-dire l'identification des positions dans le génome qui varient par rapport au génome de référence et entre les individus. Un cadre d'analyse populaire pour cette application est GATK pour le polymorphisme d'un seul nucléotide (SNP) ou les petites insertions/délétions (indels) (Figure 2). Les variantes comprenant de plus gros morceaux d'ADN (également appelées variantes structurelles) requièrent des méthodes d'appel spécifiques (cf. cet article pour une comparaison complète). Comme pour les aligneurs, je conseille de choisir le bon outil en tenant compte de facteurs clés tels que le type de variants (SNP, indel ou variants structurels), l'acceptation par la communauté, la qualité de la documentation et le nombre d'utilisateurs.
L'application la plus fréquente de RNA-seq est probablement la quantification de l'expression des gènes. Historiquement, les lectures devaient être alignées sur la séquence de référence, puis le nombre de lectures alignées sur un gène ou un transcrit donné était utilisé comme approximation pour quantifier ses niveaux d'expression. Cette approche alignement+quantification est réalisée par des outils tels que Boutons de manchette, RSEM ou comptes des caractéristiques. Cependant, cette approche a été de plus en plus dépassée par de nouvelles méthodes mises en œuvre dans des logiciels tels que Kallisto et Saumon. Conceptuellement, avec de tels outils, il n'est pas nécessaire d'aligner la séquence complète d'une lecture sur la séquence de référence. Au lieu de cela, il suffit d'aligner suffisamment de nucléotides pour être sûr qu'une lecture provient d'un transcrit donné. En d'autres termes, l'approche alignement+quantification est réduite à une seule étape. Cette approche est connue sous le nom de pseudo-mapping et augmente considérablement la vitesse de quantification de l'expression des gènes. D'un autre côté, gardez à l'esprit que le pseudo-mapping ne conviendra pas aux applications pour lesquelles l'alignement complet est nécessaire (par exemple, l'appel de variants à partir de données RNA-seq).
Le ChIP-seq est un autre exemple des différences entre les étapes d'analyse en aval et les outils requis pour les applications basées sur le séquençage. Les lectures générées par cette technique seront utilisées pour l'appel de pic, qui consiste à détecter les régions du génome présentant un excès significatif de lectures indiquant où la protéine cible est liée. Plusieurs peak callers existent et cette publication les étudie. Comme dernier exemple, je mentionnerai les données Hi-C, dans lesquelles les alignements servent d'entrée à des outils qui déterminent les matrices d'interaction et, à partir de celles-ci, les caractéristiques 3D du génome. Commenter tous les essais basés sur le séquençage dépasse le cadre de cet article (pour une liste relativement complète, voir cet article).
Avant de commencer...
La partie restante de cet article aborde des aspects qui ne sont peut-être pas strictement considérés comme des étapes de l'analyse des données HTS et qui sont largement ignorés. En revanche, je soutiens qu'il est capital que vous réfléchissiez aux questions posées en Tableau 1 avant de commencer à analyser des données HTS (ou n'importe quel type de données en fait), et j'ai écrit sur ces sujets ici et ici.
Tableau 1
| Réfléchissez-y | Action proposée |
| Avez-vous toutes les informations de votre échantillon nécessaires à l'analyse ? | Collecter systématiquement les métadonnées des expériences |
| Serez-vous capable d'identifier sans équivoque votre échantillon ? | Établir un système pour attribuer à chaque échantillon un identifiant unique |
| Où seront les données et les résultats ? | Organisation structurée et hiérarchique des données |
| Serez-vous en mesure de traiter de multiples échantillons de manière transparente ? | Extensibilité, parallélisation, configuration automatique et modularité du code. |
| Est-ce que vous ou quelqu'un d'autre sera capable de reproduire les résultats ? | Documentez votre code et vos procédures ! |
Comme mentionné ci-dessus, les données brutes HTS et certains des fichiers générés lors de leur analyse sont de l'ordre du gigaoctet, il n'est donc pas exceptionnel qu'un projet comprenant des dizaines d'échantillons nécessite des téraoctets de stockage. En outre, certaines étapes de l'analyse des données HTS sont gourmandes en ressources informatiques (par exemple, l'alignement). Cependant, l'infrastructure de stockage et de calcul requise pour l'analyse des données HTS est un élément important qui est souvent négligé ou non discuté. Par exemple, dans le cadre d'une analyse récente, nous avons examiné des dizaines d'articles publiés effectuant une analyse d'association à l'échelle du phénome (PheWAS). Les PheWAS modernes analysent 100 à 1 000 variantes génétiques et phénotypes, ce qui nécessite un stockage de données et une puissance de calcul importants. Et pourtant, pratiquement aucun des articles que nous avons examinés n'a commenté l'infrastructure nécessaire à l'analyse PheWAS. Il n'est donc pas surprenant que je vous recommande de planifier dès le départ les exigences de stockage et de calcul auxquelles vous serez confronté et de les partager avec la communauté.
Vous avez besoin d'aide pour analyser les données de séquençage de l'ADN ? Prenez contact avec freelance bioinformatics specialist et experts en génomique sur Kolabtree.
Kolabtree helps businesses worldwide hire freelance scientists and industry experts on demand. Our freelancers have helped companies publish research papers, develop products, analyze data, and more. It only takes a minute to tell us what you need done and get quotes from experts for free.

Unlock Corporate Benefits
• Secure Payment Assistance
• Onboarding Support
• Dedicated Account Manager
Sign up with your professional email to avail special advances offered against purchase orders, seamless multi-channel payments, and extended support for agreements.


