



Biologiste informaticien et le freelance Kolabtree Shaurya Jauhari writes about current challenges involved in conducting pathway analysis in bioinformatics and possible solutions to the problem.
In the contrasting era of “Imprecise Medicine” (propelling us to tune towards Precision Medicine) and inundation of biomedical data brought forth by advancements in instrumentation technologies, a lacuna persists that is largely premised over mapping data to information. The clinical experiments engender biomarqueurs (technically list of genes or genomic regions more contemporarily) that have to be expounded for their biological implications. The current suite of tools that facilitate such an endeavor is less purposeful as it neglects the ipso factoL'objectif de ce commentaire est de mettre en évidence la nature du problème, de faire la lumière sur l'organisation du génome, de réfléchir brièvement aux outils de cartographie actuels et d'imaginer des solutions possibles. Ce commentaire a pour but de mettre en évidence la nature du problème, de faire la lumière sur l'organisation du génome, de réfléchir brièvement aux outils de cartographie actuels et de conjecturer les solutions possibles.
Le diable est dans les détails
Les nombreux efforts déployés pour améliorer la résolution des données génomiques passent à côté d'un élément d'information crucial. Nous sommes impatients de voir la probabilité d'un génome $1000, mais nous nous soucions moins de l'analyse $100 000. Il existe un vaste recueil de répertoires qui contiennent les annotations des résultats expérimentaux et des cas d'une étude biologique typique. Il pourrait y avoir des définitions indiquant les implications biologiques d'un gène, ou de la voie dans laquelle ces gènes s'inscrivent, associé à une maladie. Encore une fois, ces types de magasins d'informations dynamiques ont été conservés manuellement (autrefois) et la gestion des connaissances a été reprise par des pipelines automatisés utilisant des ordinateurs et les TIC au sens large. Ces bases de données sont mises à jour en fonction d'un consensus scientifique et ont fait l'objet d'une poignée de révisions depuis leur création. Le conduit qui relie les résultats expérimentaux à leurs implications biologiques est fortement atténué, en grande partie parce que la "vraie" biologie sous-jacente est écartée.
Notre génome, d'une longueur moyenne d'environ 2 mètres, est logé dans le noyau de chacune des cellules de notre corps. En raison de la taille réduite d'une cellule et plus encore de son noyau, le génome est emballé d'une manière quelque peu tendue et molle. Cela permet à des régions du génome, plutôt éloignées d'un point de vue linéaire, de se rapprocher et d'interagir. Cet adage est grossièrement rejeté par la suite actuelle d'outils d'enrichissement (cartographie) et les résultats engendrés sont donc disproportionnés.
Les régions du génome font partie de plus grands "groupes d'action" ou voies d'accès that are technically series of chemical reactions accounting for a phenotype; healthy or diseased. When a diseased state is examined, the investigators are on the lookout for the biomarkers that have potentially gone awry and have apparently transformed the organismal body from tonique à a tressailli. Imaginez poursuivre une maladie durement combattue avec des informations mal alignées.
 Énoncé du problème - Flux de travail d'une analyse d'enrichissement typique. Il existe un certain
Énoncé du problème - Flux de travail d'une analyse d'enrichissement typique. Il existe un certain
"idiomatisme" associé à la mise en correspondance des séquences du génome avec les gènes, et qui orchestre la
les résultats en aval.
Organisation du génome
Comme nous l'avons déjà dit, le génome est suffisamment long pour être stocké linéairement à l'intérieur du noyau d'une cellule, pour chaque cellule de notre corps ou de tout autre organisme vivant. Au lieu de cela, cette entité de 2 mètres est comprimée et entassée dans une structure apparemment désordonnée, avec des boucles, des virages et des tourbillons comme on peut l'imaginer. Ce sans fin La chaîne de nucléotides ou paires de bases - Adénine (A), Cytosine (C), Thymine (T), et Guanine (G) - structure des topologies distinctes à l'intérieur du noyau tout en s'adaptant à l'exiguïté. Elles forment des boucles chromatiniennes, des compartiments/sous-compartiments, des domaines/sous-domaines qui ont une fonction, en fonction du type de cellule. (Notez que les différents types de cellules fonctionnent différemment ; une cellule nerveuse a d'autres affaires à traiter qu'une cellule musculaire ; chacune a un rôle exclusif à jouer).
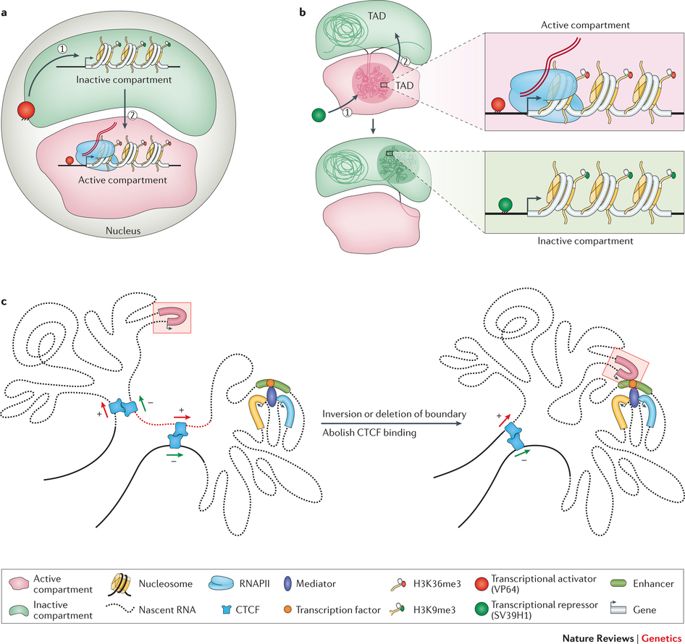
Le génome s'enroule et s'étale dans des espaces objectifs. (Crédit : https://doi.org/10.1038/nrg.
2016.112)
Bases de données sur les voies d'accès
Il existe des ontologies et des bases de données variées, dont l'encyclopédie des gènes et des génomes de Kyoto (KEGG) (https://www.genome.jp/kegg/) et Gene Ontology (http://geneontology.org/Les quelques outils que je présenterai dans la prochaine section (et qui sont souvent sujets à caution) génèrent des termes d'enrichissement qui proviennent "sélectivement" desdites bases de données. Sur la base de leurs valeurs de signification statistique, on peut déterminer s'ils représentent réellement un phénotype répertorié ou s'ils sont simplement le fruit d'un développement aléatoire (P.S. Il existe un article sur les valeurs p qui aidera probablement les profanes à comprendre l'idée de la valeur p). importance statistique. Veuillez suivre le lien https://sway.office.com/WkyHrPnVB8Ec3zPD?play à votre convenance).
Outils d'enrichissement
L'analyse d'enrichissement est un protocole computationnel de novo des régions génomiques à leurs définitions enregistrées dans les bases de données auxquelles il a été fait allusion. En ce qui concerne les outils (qui agissent comme un conduit), ils sont classiquement structurés en plusieurs catégories, à savoir l'analyse de surreprésentation (ORA), l'évaluation des classes fonctionnelles (FCS), les méthodes basées sur la topologie des voies (PT) et les méthodes d'interaction de réseau (NI).
Analyse de la surreprésentation
L'analyse de surreprésentation, via le dogme de la distribution hypergéométrique, évalue l'ensemble des gènes différentiellement exprimés pour déterminer ceux qui pourraient faire partie d'une voie biologique. Fondamentalement, un test hypergéométrique prend en compte quatre attributs pour parvenir à une décision, à savoir
- Nombre total de gènes dans l'essai considéré,
- Les gènes différentiellement exprimés,
- les gènes de la voie cible par rapport au nombre total de gènes, et
- Gènes exprimés de manière différentielle dans la voie cible.
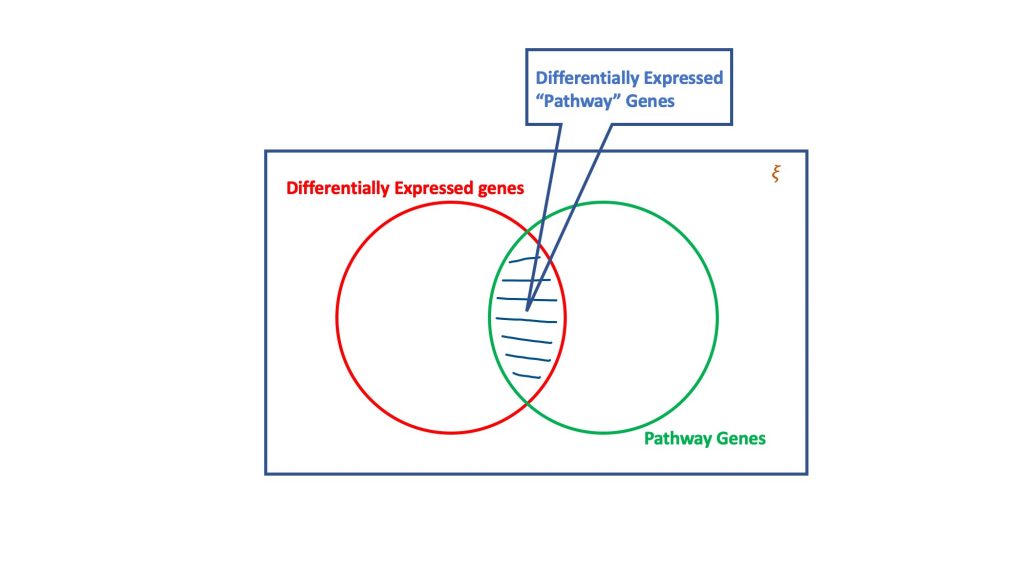
L'essence du test hypergéométrique
Despite being a simple and straightforward methodology, ORA presents its own limitations.
- Démocratie en jeu ; tous les gènes sont considérés comme égauxPourquoi est-ce un problème ? Supposons que les gènes soient filtrés sur la base de changement de plis. Nous sélectionnons les gènes dont la différence d'expression est supérieure ou égale à 2 fois (plis), dans les deux sens, négatif et positif. Bien que le minimum soit de 2 fois, ce flux de travail capture également les gènes dont les changements d'expression sont de 3 fois, 4 fois et plus. Il est certain qu'un gène dont l'expression varie de 4 fois est plus prudent qu'un gène dont l'expression varie de 2 fois. Cette manifestation est écartée par ORA.
- Ne prend en compte que les gènes les plus significatifs; Encore une fois, considérons un gène avec un fold-change de 1,9999 ou une valeur p <0,0051113 ; communément, une valeur p <0,05 est considérée comme statistiquement significative. La méthodologie ORA ne tient pas compte de ce gène dans le résultat final. Il y a clairement une perte d'information et un manque de flexibilité. (P.S. Breitling et al. ont abordé cette situation difficile en proposant une extemporanéité pour éviter les seuils. Cette révision utilise une approche itérative qui ajoute un gène à la fois pour compiler un ensemble de gènes pour lesquels une voie est significative de manière optimale).
- Aucun gène ne fonctionne de manière isoléeCela découle des limites susmentionnées : en traitant le gène comme une entité indépendante, on perd l'essentiel de la contribution polygénique à un phénotype. L'un des objectifs de l'analyse de l'expression génétique pourrait être d'élucider les cohortes de gènes dont les profils d'expression sont congruents. Cette symphonie met en évidence des gènes fonctionnellement apparentés ou des gènes travaillant à un état biologique commun.
- Voies mutuellement indépendantes; ORA suppose également que les voies ne fonctionnent pas en tandem (ou en succession). Cette hypothèse est principalement erronée, car des séries de réactions chimiques peuvent très bien se précéder ou se succéder.
Evaluation de la classe fonctionnelle
Contrary to ORA, FCS methods subsume all the contextual genes as well as their association statistiques (fold-change, p-value) and compute a en cours d'exécution score d'enrichissement pour les groupements de gènes (basé sur certaines connaissances fonctionnelles comme l'ontologie des gènes ou les voies KEGG), par exemple GSEA du Broad Institute (http://software.broadinstitute.org/gsea/index.jsp). Une exécution typique de FCS analyserait le changement d'expression de l'ensemble des gènes dans la liste (pas de classement par signification statistique ou autre) des gènes différentiellement exprimés dans une expérience. Le résultat principal de l'analyse d'enrichissement des ensembles de gènes est un score d'enrichissement (ES) qui reflète le degré de surreprésentation d'un ensemble de gènes en haut ou en bas d'une liste classée de gènes ; pourquoi en haut et en bas ? parce que ce sont les gènes les plus éloignés de la normale, en termes de changement d'expression. Un score ES positif pour un ensemble de gènes (ou une voie cible), GS) seront indicatifs des gènes de la liste (GL) se situant dans le haut (les plus régulés vers le haut ; 1,2,3 ...), tandis qu'un score ES négatif signifie que les gènes composants se situent dans le bas (les plus dérégulés ; n-3, n-2, n-1, n, où n est le nombre total de gènes). P.S. L'ES devient l'ES normalisé (NES) lorsqu'on corrige le problème des tests multiples (taux de fausse découverte, par ex. Méthode Bonferroni).
En résumé, les méthodes FCS sont nettement meilleures que les méthodes ORA,
- en évitant l'exigence d'un seuil arbitraire pour classer les gènes comme significatifs ou non significatifs.
- apprécier les informations sur l'expression des gènes pour suivre les changements systématiques dans la voie ; cela permet de rendre compte de l'interdépendance des gènes.
Cependant, les méthodes de CSC présentent également certaines lacunes.
- Les voies étant analysées indépendamment, les gènes qui régulent plusieurs voies peuvent ne pas être comptabilisés.
- De nombreuses méthodes de SFC classent les gènes d'une liste sur la base des changements dans l'expression des gènes. Un scénario dans lequel la différence de classement reflète une variance inégale (et peut-être exponentielle) de l'expression pourrait peut-être constituer une mesure injuste.
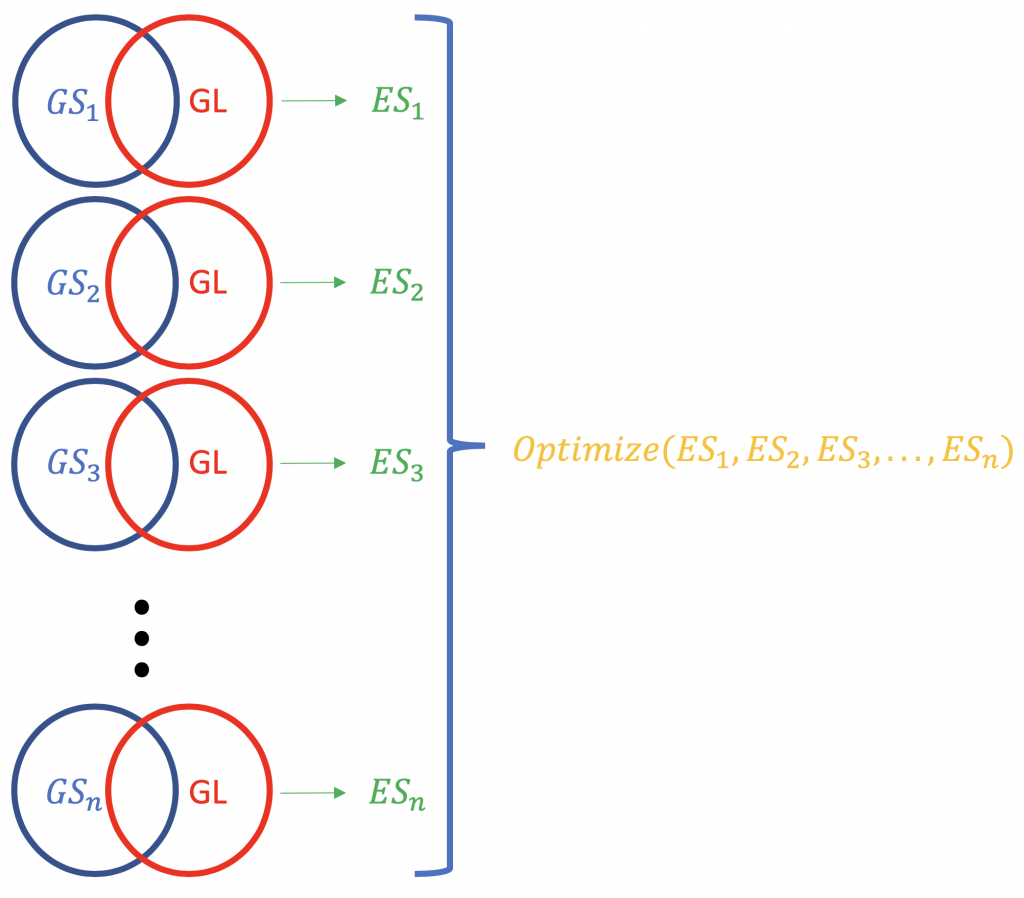
La normalisation des scores ES relatifs à une liste de gènes.
Approches basées sur la topologie des voies
Une lacune cruciale des méthodes ORA et FCS est qu'elles ignorent la structure des voies. L'ordre des gènes qui sont régulés dans une voie est essentiel pour retracer les effets causaux. Naturellement, il pourrait y avoir exactement deux voies avec les mêmes composants génétiques, mais la hiérarchie d'activation pourrait être entièrement différente. Si c'était le cas, les méthodes ORA/FCS auraient abouti à des termes d'enrichissement similaires. C'est un problème. Les méthodes Pathway Topology(PT) supposent l'exclusivité de la fonction en fonction des interactions spécifiques, ce qui est également conforme à la logique générale. Des exemples d'outils sont SPIA (https://www.bioconductor.org/packages/release/bioc/manuals/SPIA/man/SPIA.pdf), GGEA, et PARADIGM. En général, les outils de cette catégorie ont un score local et un score global. Le score local au niveau du gène doit calibrer les changements de plis dans l'expression du gène et des gènes en amont, tandis que le score global doit évaluer la mesure au niveau de la voie pour la relation avec l'ensemble des gènes. Néanmoins, cela signifie également que les méthodes de PT surajustent les données pour une condition/un type de cellule particulier.
Analyse basée sur l'interaction des réseaux
Il s'agit plutôt d'une catégorie sous-estimée qui est encore peu mise en œuvre, malgré sa formulation datée. Des méthodes comme EnrichNet , NetPEA (http://www.dx.doi.org/10.1109/BIBM.2013.6732493) have been proposed close to a decade ago, but they haven’t gained much traction because of limited tools available. This facade constraints the dexterity of the theme as no improvements are documented. That turns out to be an open-ended recherche problem.
L'apparent Panacée
Vous avez probablement maintenant une idée de l'enrichissement fondamental/l'analyse des voies de transmission et du type d'outils qui l'aident. Cependant, comme je l'ai déjà mentionné, tous les outils disponibles (appartenant aux catégories marquées) reposent sur un paramètre biaisé d'une fenêtre linéaire à travers la région interrogée. Si les segments intermédiaires du génome entrent dans ce cadre, ils sont répertoriés en tant que enrichiou non. L'essentiel est de disposer d'un outil qui, peut-être, permet de saisir une centre et diamètre d'un cercle hypothétique, en quelque sorte pour mettre en évidence l'organisation 3D basée sur les interactions du génome pour la région.

GREAT offre plusieurs options avancées pour spécifier la dimension linéaire autour du site de début de transcription d'un gène.
 Enrichr présente une sélection simple du type de génome et du nombre de gènes dans la région linéaire présumée.
Enrichr présente une sélection simple du type de génome et du nombre de gènes dans la région linéaire présumée.
Comme le montre la figure 4, il n'y a aucune appréciation de la "véritable" organisation spatiale du génome. Il s'agit d'un problème et d'une profonde faille qui persiste dans le domaine actuel de l'analyse de l'enrichissement. Néanmoins, il existe également une pertinence flagrante de l'analyse d'enrichissement. usines de transcriptionLes usines de transcription, identifiées comme des sites dans l'espace nucléaire qui attirent des éléments régulateurs distants pour "faire la fête en interne". En plaisantant, je remarque souvent qu'à l'instar de ce que dirait quelqu'un qui est furieux contre quelqu'un ou quelque chose, les usines de transcription (personnifiées) pourraient réprimander le génome... ".La transcription se fera sur mon cadavre, pas autrement !". Le thème voisin des usines de transcription fait l'objet d'une discussion future adjacente. Cependant, il matérialise le dogme des interactions cis-régulatrices qui fait défaut dans la pratique contemporaine.
Pour conclure, je voudrais faire remarquer que l'analyse des voies de communication est une fraction cruciale, et souvent négligée, de l'analyse de l'environnement. Bioinformatique pipeline. Il est toujours possible d'adapter les méthodologies existantes en fonction des données génomiques, qui évoluent en ce moment même. Lorsque nous aurons accès à un volume croissant de données, nous serons confrontés non seulement à un problème d'infrastructure, mais aussi à un problème d'algorithme.
Besoin de engager un consultant en bioinformatique? Travaillez avec des scientifiques indépendants sur Kolabtree. C'est gratuit de poster votre projet et d'obtenir des devis d'experts.
Experts connexes :
Pigiste en bioinformatique | Génétique végétale | Biologie du développement | Thérapie génique | Cellules souches |
Analyse des données de séquençage de l'ADN |Génétique animale | Interactions médicamenteuses | Génétique et génomique
Kolabtree helps businesses worldwide hire freelance scientists and industry experts on demand. Our freelancers have helped companies publish research papers, develop products, analyze data, and more. It only takes a minute to tell us what you need done and get quotes from experts for free.

Unlock Corporate Benefits
• Secure Payment Assistance
• Onboarding Support
• Dedicated Account Manager
Sign up with your professional email to avail special advances offered against purchase orders, seamless multi-channel payments, and extended support for agreements.


