



Sundeep Agarwal, dispositivo médico regulatory consultant on Kolabtree, shares the essential requirements of a FDA 510k premarket notification to ensure success.
Con la actual $156 de tamaño de mercado, y se espera que $208 mil millones [1] by 2023, the US medical device market is undoubtedly lucrative. Additionally, the aging population, history of chronic diseases, the salud system and disruption in supply chain encourages global manufacturer to invest and expand their business horizon in the United States. Like any other global manufacturer, if you think you can leave an imprint in the world largest medical device market, you have just hit on the right blog to prepare and understand the regulatory requirement with respect to FDA’s 510(k)for your device which is utmost vital for the application process.
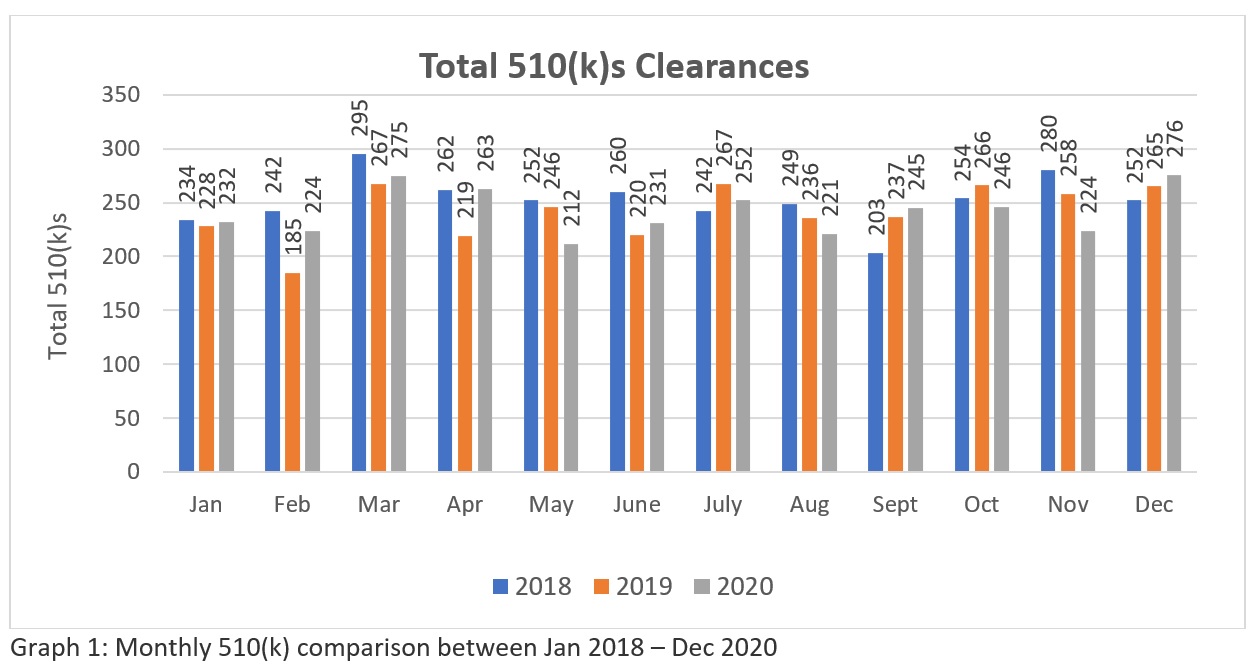
A trend analysis of three years FDA’s 510k clearances data (Source: https://www.fda.gov/medical-devices/device-approvals-denials-and-clearances/510k-clearances) revelan que un total de 3025-510(k)swere autorizada entre enero de 2018 y diciembre de 2018, 2894 - 510(k) entre enero de 2019 y diciembre de 2019 y 2901 - 510(k) autorizados entre enero y diciembre de 2020. Estas cifras (véase la tabla 1) sugieren que, en la actualidad, la FDA tiene el potencial de autorizar más de doscientos 510(k) al mes para que la industria satisfaga las necesidades del mercado sanitario en los Estados Unidos. La representación gráfica (véase el Gráfico 1) indica la coherencia de la FDA en su enfoque de revisión en medio de la pandemia mundial. Aunque el número no es significativamente grande en comparación con el gran número de solicitudes presentadas a nivel mundial. Esperamos que la FDA aumente sus recursos en el futuro. Por lo tanto, los fabricantes necesitan una preparación y presentación cuidadosa para asegurarse de que pasan en su primer intento.
| Año 2018 | Año 2019 | Año 2020 | |
| Total de 510(k) | 3025 | 2894 | 2901 |
| Total con resúmenes | 2885 | 2735 | 2759 |
| Total con declaraciones | 140 | 159 | 142 |
Tabla 1: La tabla indica los 510(k) anuales autorizados entre enero de 2018 y diciembre de 2020
Requisitos FDA 510k: Cómo encontrar las claves
A no ser que se conozca bien la normativa de la FDA Preparación y presentación del 510(k) proceso, puede ser realmente estresante y desafiante. Si un fabricante de productos tiene la intención de comercializar un producto en los EE.UU. que es de clase I, II y III, pero no un exento de un 510(k) o que no no requiere una solicitud de aprobación previa a la comercialización (PMA), entonces es elegible para una 510(K). Además, el fabricante debe tener claros los términos "producto anterior" y "equivalencia sustancial (SE)". Un producto comercializado legalmente en los EE.UU. para el que un fabricante quiere reclamar una equivalencia se conoce generalmente como producto anterior. Por otro lado, la equivalencia sustancial significa establecer una comparación basada en pruebas para que un nuevo producto que un fabricante quiere colocar en el mercado de EE.UU. sea seguro y eficaz como el producto predicado existente, en particular con respecto al mismo uso previsto y las características tecnológicas. En caso de que el nuevo producto difiera en sus características tecnológicas, no debería diferir en términos de seguridad y eficacia y el fabricante puede demostrar que el producto es tan seguro y eficaz como el producto existente comercializado legalmente.
Desbloquear el proceso
Algunos de los factores fundamentales para el éxito de un 510(k) son la correcta clasificación del código del producto, la identificación y disponibilidad de un dispositivo precedente en el mercado estadounidense, una comparación de equivalencia sustancial bien planificada con pruebas, un sistema de gestión de la calidad sólido, controles de diseño, el estricto cumplimiento de los formularios actuales de la FDA y la mejor utilización de la lista de comprobación de denegación de aceptación (RTA) (consulte también la figura 1). La lista de comprobación de denegación de aceptación (RTA) orienta sobre los diversos criterios de aceptación que sigue la FDA al realizar una revisión sustantiva de un 510(k). Para los tres tipos de 510(k), hay diferentes tipos de RTA disponibles. Se puede acceder a la lista de comprobación RTA desde aquí.
El fabricante debe tener en cuenta que no es el número de páginas o el mero hecho de presentar los resultados de las pruebas o los estudios lo que les haría obtener un Autorización 510(k) En lugar de ello, una redacción secuencial bien explicada (como recomienda la FDA en su documento de orientación) basada en la justificación científica y las pruebas que serán revisadas cuidadosamente por la FDA dentro de un plazo fijo consigue en última instancia el 510(k). La mayoría de los 510(k) se rechazan por falta de claridad o por pruebas inadecuadas para reclamar una equivalencia sustancial [como en el 510(k) tradicional] o por una comparación insuficiente con las normas de conformidad reconocidas por la FDA [como en el 510(k) abreviado]. Se observa que la falta de gestión de los controles de diseño y la ausencia de un sistema de calidad bien establecido es otra de las grandes razones de este fracaso. Por último, la falta de disponibilidad de un equipo competente o la falta de comprensión clara de la normativa es un factor innegable que contribuye al rechazo, además de los anteriores.

Figura 1: Puntos clave a tener en cuenta para una notificación previa a la comercialización 510(k)
Tipos de notificación previa a la comercialización de la FDA 510(k)
En general, hay tres tipos de 510(k) que un fabricante puede presentar a la FDA (véase la figura 2). Estos son:
(1) Tradicional 510(k) - La mayoría de las solicitudes 510(k) son de este tipo,
(2) Especial 510(k) - Sólo se requiere cuando se realizan cambios en la etiqueta o el diseño o ciertos cambios en la indicación de uso en un producto previamente autorizado. El contenido debe cumplir los requisitos definidos en 21 CFR Parte 807.87 y 21 Parte 807.90,
(3) Abreviado 510(k) - Se aplica en el caso de que un fabricante pueda elaborar informes sobre el uso de controles especiales u orientaciones o una declaración de conformidad basada en las normas reconocidas por la FDA.

Figura 2: Tipos de notificaciones previas a la comercialización 510(k)
El proceso de presentación de 510k
Antes de una presentación, el fabricante tendrá que registrar su organización en la FDA. El proceso se denomina registro de establecimiento según el 21 CFR Parte 807 tras el pago de las tasas directamente a la FDA y se renovará anualmente. Para el actual ejercicio 2021, la tasa es de $ 5.546 para el registro de un establecimiento. Una vez que se comprueben las tasas exactas visitando el sitio web oficial de los programas de tasas de usuario de la FDA.
La FDA recomienda 20 secciones en un 510(k) tradicional o abreviado, pero no necesariamente todas las secciones serán aplicables a un fabricante. A veces, si una información en una sección particular no se aplica a su dispositivo, pueden incluir el título de la sección y escribir "Esta sección no se aplica" o "N/A" bajo la misma. A continuación se enumeran las secciones principales recomendadas en el documento de orientación de la FDA para el 510(k):
- Hoja de presentación de la tasa de usuario de dispositivos médicos (formulario FDA 3601): Indica el recibo de una tasa de usuario pagada a la FDA por el fabricante.
- Hoja de presentación de la revisión previa a la comercialización del Centro de Dispositivos y Salud Radiológica (CDRH) (Formulario FDA 3514): Se trata de un formulario voluntario para proporcionar todo tipo de información administrativa a la FDA sobre la organización y la presentación.
- Una carta de presentación del 510(k): En esta carta debe incorporarse una descripción sobre el propósito, el contenido y la información administrativa sobre el 510(k). Se recomienda consultar el Apéndice A del "Formato para los 510(k) tradicionales y abreviados - Guía para la industria y el personal de la Administración de Alimentos y Medicamentos; dtd 13 de septiembre de 2019".
- Declaración de indicaciones de uso (formulario FDA 3881): Debe ser uniforme en todo el 510(k). También debe definir si el dispositivo se va a comercializar como de uso con receta o de venta libre (OTC).
- Resumen de 510(k) o Declaración de 510(k): Debe prepararse de acuerdo con el 21 CFR parte 807. Aquí se espera que el fabricante resuma el 510(k) e incorpore información sobre el resto del contenido.
- Declaración de veracidad y exactitud: Es una declaración de una persona autorizada de la organización que certifica que toda la información presentada a la FDA relacionada con el 510(k) es veraz y exacta.
- Resumen y certificación de la clase III: Sólo es aplicable a los productos de la clase III. Es un resumen de la seguridad y la eficacia y una garantía de que se ha realizado una búsqueda razonable y el fabricante dispone de toda la información de seguridad pertinente basada en dispositivos similares comercializados.
- Certificación o declaración financiera: Si un fabricante presenta pruebas clínicas, deberá adjuntar una declaración de divulgación del investigador clínico. Se puede remitir el formulario 3454 o el formulario 3455 de la FDA.
- Declaraciones de conformidad e informes de síntesis: Aquí se proporcionará información relacionada con el uso de normas de consenso voluntario o la base del uso general de dichas normas.
- Descripción del dispositivo: Se debe incluir una breve descripción del diseño del dispositivo, los modelos o los accesorios.
- Resumen ejecutivo/Comparación de predicados: En esta sección se recomienda una breve descripción del dispositivo, las indicaciones de uso y la tecnología junto con una tabla comparativa de dispositivos.
- Discusión sobre la equivalencia sustancial: Una comparación detallada entre el dispositivo del fabricante y el dispositivo predicado para demostrar la equivalencia sustancial.
- Propuesta de etiquetado: Incluirá el etiquetado propuesto para el producto sanitario según el 21 CFR 807.87(e) o según los requisitos del 21 CFR 809.10 en el caso de un producto de diagnóstico in vitro.
- Esterilización y vida útil: El método de esterilización, la validación pertinente y la vida útil declarada deben incluirse en esta sección.
- Biocompatibilidad: Protocolo de estudios, informes y garantía de que los estudios de biocompatibilidad se han realizado siguiendo las buenas prácticas de laboratorio. La FDA recomienda el uso de la norma ISO 10993 para los estudios de biocompatibilidad.
- Software: Si el dispositivo incorpora un programa informático, se aplicará esta sección.
- Compatibilidad electromagnética y seguridad eléctrica: Aplicable principalmente a los dispositivos eléctricos o activos. La FDA recomienda el uso de la norma ANSI/AAMI (ES) 60601-1 para las pruebas generales de seguridad o un método equivalente.
- Pruebas de rendimiento - Banco: Se recomienda incluir las distintas pruebas de rendimiento realizadas por el fabricante o en un laboratorio de terceros, que pueden incluir, entre otros, resultados de pruebas mecánicas o de ingeniería o biológicas.
- Pruebas de rendimiento - Animales: Si se realizaron estudios en animales y se incluyen en la presentación, la FDA recomienda describir las pruebas y proporcionar los resultados que apoyan las características de rendimiento.
- Pruebas de rendimiento - Clínicas: Si la presentación incluye datos/estudios clínicos, la FDA espera que se incluya información sobre el protocolo y el objetivo del estudio clínico, los métodos de prueba, los criterios de valoración del estudio y las herramientas estadísticas utilizadas en el estudio clínico.
Por favor, tenga en cuenta que no hay una plantilla estándar o un formato de solicitud 510(k) listo para rellenar; aunque el 21 CFR Parte 807, Subparte E de la FDA describe el procedimiento y guía al fabricante sobre el registro del establecimiento y el listado de dispositivos. Además, se pueden descargar varios formularios relevantes asociados a dicha presentación que son útiles para preparar desde el enlace oficial de la FDA, es decir https://www.fda.gov/medical-devices/premarket-notification-510k/510k-forms . Es aconsejable consultar las diversas guías de la FDA sobre el 510(k), disponibles en el dominio público, para cumplir con los requisitos reglamentarios y documentar en consecuencia. Aunque no es una práctica para llevar a cabo las inspecciones de las instalaciones de 510(k) antes de la autorización, pero el fabricante debe implementar un sistema de calidad robusto según el requisito 21 CFR Parte 820 y estar listo para la inspección, en caso de que se lleve a cabo.
También existe una disposición conocida como "Programa de revisión por terceros" para determinados dispositivos de riesgo bajo o moderado. Como su nombre indica, no es la FDA la que revisa directamente la solicitud 510(k), sino que lo hace una organización tercera acreditada y aprobada por la FDA. Sobre la base de la revisión y la recomendación realizada por la tercera parte acreditada, la FDA toma una decisión sobre la autorización 510(k). En la actualidad existen 10 organizaciones de terceros de este tipo. El remitente o el fabricante debe comprobar si su dispositivo puede ser revisado por una tercera parte en https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfpcd/classification.cfm
Calendario de la solicitud 510k
Un remitente o fabricante al presentar un 510(k) a la FDA también debe preparar una copia electrónica de su 510(k) y presentarla al CDRH o al CBER en el centro de control de documentos (DCC). Sobre la base de la información presentada, se pedirá al fabricante información adicional durante las diferentes etapas de revisión (véase la figura 3).
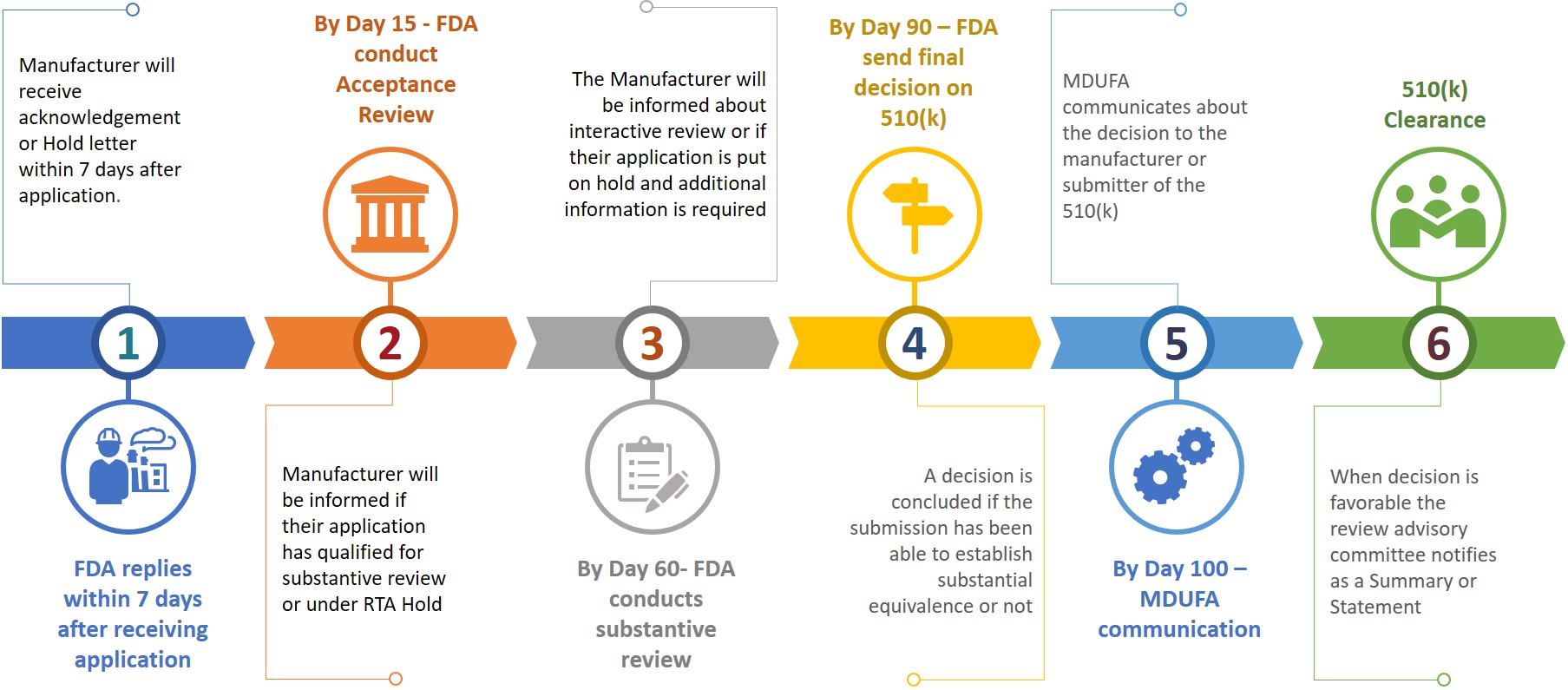
Figura 3: Cronograma tentativo de revisión
El remitente dispondrá de 180 días naturales para responder cuando reciba una retención de RTA o si la FDA desea información adicional. Si no se resuelve ningún problema en los 18 días naturales permitidos, se eliminará automáticamente del sistema de revisión o se considerará retirado. En caso de eliminación o retirada de un 510(k), el remitente tendrá que volver a presentar una nueva solicitud tras pagar la tasa necesaria, mientras que el número K puede citarse en la nueva solicitud para el mismo dispositivo. La autorización 510(k) puede obtenerse en un plazo de 100 días después de la presentación, mientras que la autorización puede tardar entre 6 y 9 meses.
Referencias
- El programa 510(k): Evaluación de la equivalencia sustancial en las notificaciones previas a la comercialización [510(k)] Guía para la industria y el personal de la Administración de Alimentos y Medicamentos Documento publicado el: 28 de julio de 2014.
- Formato para los 510(k) tradicionales y abreviados Orientación para la industria y el personal de la Administración de Alimentos y Medicamentos; Documento publicado el 13 de septiembre de 2019.
- [1]El panorama de la tecnología médica (EE.UU.), preparado en colaboración con la Unidad de Industria y Análisis (I&A) de la Administración de Comercio Internacional https://www.selectusa.gov/medical-technology-industry-united-states (Consultado por última vez el 11 de junio de 2021)

Sobre el autor
Sundeep Agarwal, Experto en la materia y consultor de la FDA, CE (MDR e IVDR)
With a decade of experience, he is globally sought-after Leader, Speaker & Consultant in the field of QA & RA, Quality Management System, Product Design & Development, Gestión de riesgos, Commercial Scale-up, Industrial Manufacturing and Clinical Studies of medical devices.An active member of a Technical Group (Software as Medical Device) at Asian Harmonization Working Party.He joins Medical Device industry/government, collaborated conferences a speaker and panelist frequently on ISO 13485, EU MDR, IVDR, CE Certification, CER, PMS, USFDA, 510(K), ISO 14971, MDSAP, Combination Devices, Inteligencia Artificial , etc. He prominently serves as a guest lecturer in various MBA and Pharmacy educational institutions in India. Póngase en contacto con él directamente para un proyecto en Kolabtree.
Kolabtree helps businesses worldwide hire freelance scientists and industry experts on demand. Our freelancers have helped companies publish research papers, develop products, analyze data, and more. It only takes a minute to tell us what you need done and get quotes from experts for free.

Unlock Corporate Benefits
• Secure Payment Assistance
• Onboarding Support
• Dedicated Account Manager
Sign up with your professional email to avail special advances offered against purchase orders, seamless multi-channel payments, and extended support for agreements.


