



Shreya Chenni, redactor autónomo de normativa for medical devices, provides a 10-minute guide to FDA design controls for your dispositivo médico.
Una de las principales causas de las retiradas de productos sanitarios es la falta de control del diseño, tal y como identificó la FDA [3,3a]. Los controles de preproducción se añadieron entonces a la normativa sobre prácticas correctas de fabricación de dispositivos. Los controles de diseño son un conjunto interrelacionado de prácticas y procedimientos que se incorporan al proceso de diseño y desarrollo, es decir, un sistema de controles y equilibrios. Los controles de diseño aumentan la probabilidad de que el diseño transferido a la producción se traduzca en un dispositivo adecuado para su uso previsto.
Aplicabilidad
Todos los dispositivos de clase II, III y los siguientes de clase I están sujetos a controles de diseño:
- Devices automated with computer software
- 868.6810 Sonda, aspiración traqueobronquial
- 878.4460 Guante de cirujano
- 880.6760 Sujeción, protección
- 892.5650 Sistema, aplicador, radionúclido, manual
- 892.5740 Fuente, teleterapia de radionúclidos
- Dispositivos automatizados con programas informáticos
- Catéteres de aspiración traqueobronquial
- Guantes de cirujano
- Sujeciones de protección
- Sistema, radionúclido, aplicador, manual
- Fuente, teleterapia con radionúclidos
Design controls apply to all D&D activities – for novel or improved devices being developed in the pre-market phase, as well as for changes to existing, marketed devices. Design controls do not apply to investigación activities conducted during the proof of concept stage.
Design controls can be applied to any desarrollo de productos process. The following flowcharts are examples of the design controls applied to a traditional Waterfall design process and V-model process for Software (SW).
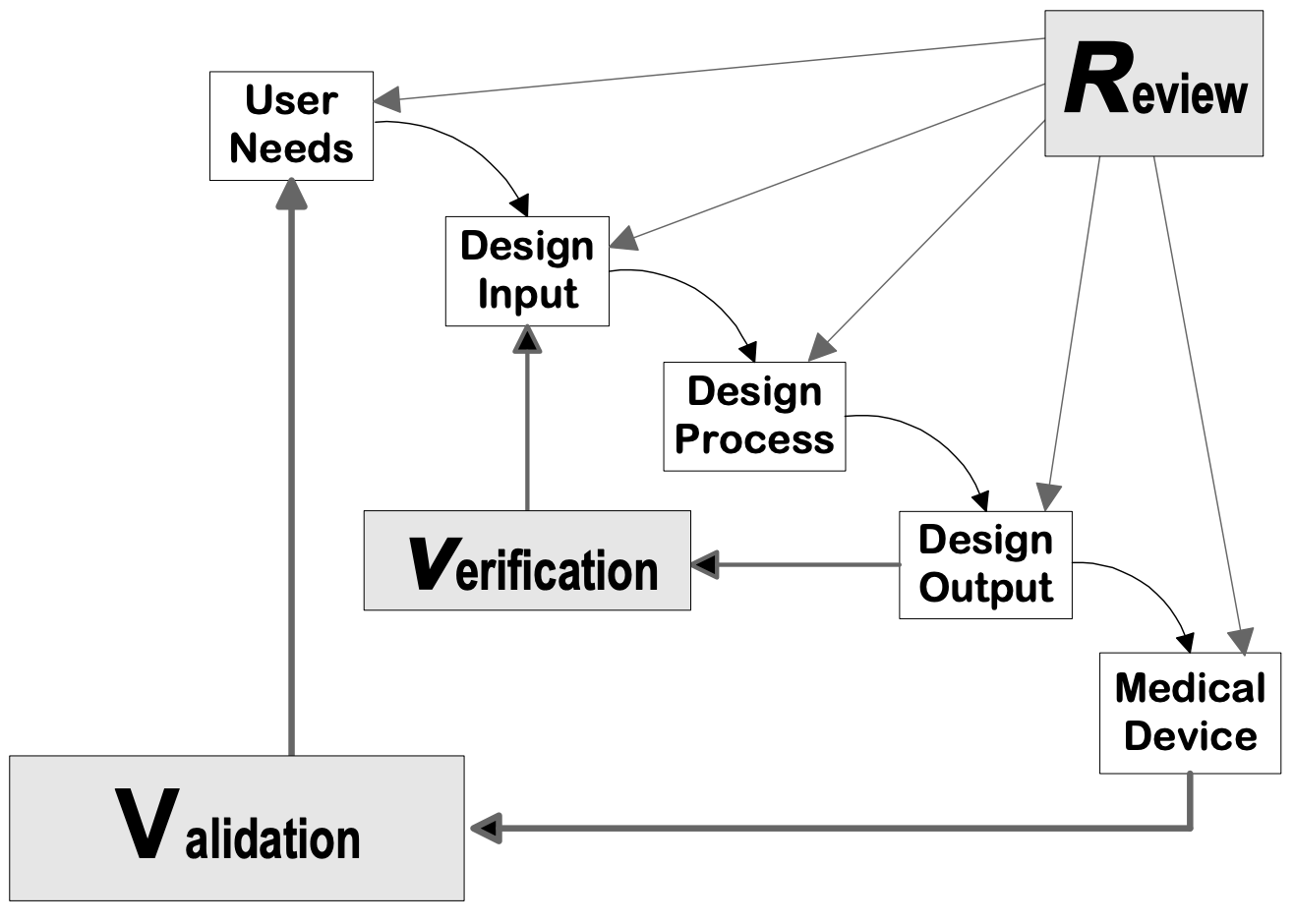
Fuente: Guía de control de diseño para fabricantes de dispositivos médicos, CDRH-FDA
Fases de diseño del dispositivo
Fase de planificación de D&D
Deben establecerse y mantenerse planes de D&D. El plan describirá o hará referencia a las actividades de diseño y desarrollo y asignará las responsabilidades para su aplicación. Como mínimo, la FDA recomienda incluir lo siguiente en el plan:
- Metas y objetivos del programa D&D
- Delimitación de las responsabilidades de las actividades de diseño
- Identificar las tareas principales, los resultados y asignar la responsabilidad de cada tarea
- Programación de las principales tareas de acuerdo con el calendario principal de desarrollo
- Identificar las principales revisiones y puntos de decisión
- Identificar a los revisores, al equipo revisor y los procedimientos que deben seguir los revisores
- Controles de la documentación de diseño
- Actividades de notificación
El plan debe ser revisado, actualizado y aprobado a medida que el diseño y el desarrollo evolucionan.
Fase de entrada del diseño
This is the starting point for product design. The medical device is designed and developed to meet the user requirements. Gather the user requirements from various sources such as customer surveys, feedback from the physicians, complaints. These requirements are transferred into design inputs.
- Entradas de diseño: Son los requisitos físicos y de rendimiento de un dispositivo que se utilizan para su diseño. La fase de entrada del diseño consiste en convertir los requisitos del usuario en requisitos del producto. Los requisitos reglamentarios se tienen en cuenta a la hora de definir las entradas de diseño. Los requisitos de entrada del diseño deben ser exhaustivos, inequívocos y verificables objetivamente. Los requisitos de entrada pueden agruparse en 3 categorías:
- Requisitos funcionales, que describen lo que hace el dispositivo. Por ejemplo: La silla de ruedas se moverá hacia delante cuando lo indique el usuario
- Los requisitos de rendimiento, que especificarán cuánto y cómo debe funcionar el dispositivo. Por ejemplo: La silla de ruedas se desplazará con una velocidad de 2 m/s en la dirección de avance
- Los requisitos de interfaz, especifican las características del dispositivo que son fundamentales para la compatibilidad con los sistemas externos, como la interfaz usuario/paciente. Por ejemplo: La silla de ruedas tendrá botones con símbolos indicadores de dirección
- Este es un ejemplo, que define las necesidades del usuario y las convierte en entradas de diseño:
- Necesidad del usuario
El dispositivo debe ser portátil y tener bluetooth
- Necesidad del usuario
-
- Aportación de diseño
Identifique la norma aplicable reconocida por la FDA o una norma internacional que deba cumplir. Por ejemplo:
-
-
- IEEE ANSI C63.27-2017 Norma nacional americana para la evaluación de la coexistencia inalámbrica
- AAMI TIR 69: Association for the Advancement of Medical Instrumentation – Gestión de riesgos of Radio-frequency Wireless Coexistence for Medical Devices and Systems (2017)
- IEC 60601-1-2 Edición 3: 2007: Equipos Eléctricos Médicos - Parte 1-2: Requisitos generales de seguridad - Norma colateral: Compatibilidad electromagnética - Requisitos y ensayos
- UL 2054 - Norma para pilas domésticas y comerciales
-
Enumera el rendimiento y otras aportaciones específicas. Por ejemplo:
-
-
- El dispositivo deberá ser alimentado por corriente continua
- Se utilizará el módulo Bluetooth
- Peso: aprox. 6lbs o 6lbs+/- 2lbs (Las entradas deben tener límites cuantitativos para garantizar la verificación)
-
Fase de diseño de salida
Los productos de diseño son los resultados de un esfuerzo de diseño en cada fase de diseño y al final del esfuerzo total de diseño. Ejemplos de resultados del diseño son los planos de ingeniería, el etiquetado, las instrucciones de trabajo y otras especificaciones del producto. Otros productos de diseño son los resultados del análisis de riesgos, los resultados de las actividades de verificación, los resultados de las pruebas de biocompatibilidad y el código fuente del software.
Los resultados del diseño no deben ser liberados antes de ser revisados y aprobados por el personal responsable. También hay que tener en cuenta que cualquier cambio en el dispositivo después de la aprobación de las salidas/entradas del diseño se controlará mediante la revisión y aprobación por parte del personal correspondiente. Al final de esta fase se requiere una revisión del diseño.
Fase de revisión del diseño
Después de la fase de salida del diseño debe realizarse una revisión del mismo. Las revisiones de diseño deben seguir los procedimientos establecidos y documentarse en el Archivo Histórico de Diseño (DHF). Los participantes de cada revisión del diseño deben tener representantes de todos los grupos funcionales.
Se recomienda realizar revisiones formales al final de los hitos importantes del proyecto. Normalmente, las revisiones del diseño se realizan después de la fase de salida del diseño, la fase de V&V y la fase de transferencia del diseño. Esto también depende de la complejidad del desarrollo del dispositivo. La FDA exige al menos una revisión del diseño.
Fase de verificación del diseño
La verificación del diseño es la confirmación mediante pruebas objetivas de que el resultado del diseño se ajusta a los datos de entrada del mismo. Básicamente, se trata de un diseño de entrada = diseño de salida. Las actividades de verificación deben realizarse según los procedimientos establecidos. Algunos ejemplos de verificación son las pruebas eléctricas y de compatibilidad electromagnética, la inspección visual, las actividades de pruebas no clínicas, el análisis del árbol de fallos del proceso o del diseño y el análisis de modos de fallo y efectos. La verificación garantiza el cumplimiento de los requisitos técnicos de las especificaciones del producto. Todas las actividades de verificación deben estar documentadas.
Matriz de trazabilidad: Este documento consta de entradas y salidas de diseño enumeradas en un formato tabular. Para cada entrada se hace referencia a la salida correspondiente. Este método de verificación se utiliza cuando las entradas y salidas son ambos documentos.
Fase de validación del diseño
La validación del diseño consiste en establecer mediante pruebas objetivas que las especificaciones (requisitos especificados) se ajustan a las necesidades del usuario y al uso o usos previstos.
- La validación de procesos significa establecer mediante pruebas objetivas que un proceso produce de forma consistente un resultado o producto que cumple con sus especificaciones predeterminadas.
- Validación del diseño: establecer mediante pruebas objetivas que las especificaciones del producto se ajustan a las necesidades del usuario y al uso o usos previstos.
La validación suele ser realizadas en condiciones reales o simuladas. Examples of validation include ensayos clínicos, la evaluación clínica, las pruebas de factores humanos, el envasado y etiquetado de direcciones, los análisis y las inspecciones. Deben documentarse los resultados de las actividades de validación y/o los informes de validación, que formarán parte del Archivo Histórico de Diseño (DHF).
Fase de transferencia del diseño
Una vez finalizada la fase de V&V, se produce la transferencia del diseño. Esto incluye la transferencia del diseño del dispositivo a las especificaciones del producto garantizando la calidad del mismo. Esta fase es muy crítica, ya que una vez que se inicie la producción del dispositivo, éste será sometido a un control de cambios de diseño y puede dar lugar a pérdidas financieras si se encuentran problemas. La transferencia del diseño debe realizarse según el procedimiento establecido. Además, hay que asegurarse de que los documentos que incluyen las especificaciones del producto sean revisados y aprobados antes de iniciar la transferencia del diseño.
Fase de cambios de diseño
El control de los cambios de diseño comienza con la transferencia del diseño y continúa durante todo el ciclo de vida. Cualquier cambio de diseño después de la transferencia de diseño dará lugar a un Aviso de Cambio de Ingeniería (ECN) que se llevará a cabo según un procedimiento establecido. Hay que asegurarse de que, con cualquier cambio de diseño, los documentos relacionados, como el informe de gestión de riesgos, las instrucciones de uso y los informes de verificación y validación, se revisen y actualicen también.
Archivo histórico de diseño (DHF)
El DHF es específico de la FDA estadounidense. La norma ISO 13486:2016 no exige que el fabricante mantenga un DHF.
Para cada proyecto se mantiene un archivo de historial de diseño que incluye todos los entregables de cada fase. Incluye la información más reciente del producto. Los documentos de diseño y desarrollo deberán estar fácilmente disponibles y ser accesibles cuando se necesiten. Los contratos de diseño y desarrollo deben especificar explícitamente el derecho del fabricante a la información de diseño y establecer normas sobre la forma y el contenido de la documentación de diseño.
En la práctica, los controles de diseño proporcionan a los gestores y diseñadores una mayor visibilidad del proceso de diseño. Con una mayor visibilidad, los gestores pueden dirigir el proceso de diseño de forma más eficaz, es decir, reconocer antes los problemas, hacer correcciones y ajustar la asignación de recursos. Los diseñadores se benefician tanto de una mayor comprensión del grado de conformidad de un diseño con las necesidades de los usuarios y los pacientes, como de una mejor comunicación y coordinación entre todos los participantes en el proceso.
¿Necesita ayuda para entender e implementar los controles de diseño de la FDA para su dispositivo médico? Póngase en contacto con expertos consultores de dispositivos médicos en Kolabtree.
Referencias:
- FDA, Guía de control de diseño para fabricantes de productos sanitarios
- Prácticas de regulación médica, una perspectiva internacional, Val Theisz
- Control de diseño, presentación de Joseph Tartal
3a. Registro Federal / Vol. 61, No. 195
Kolabtree helps businesses worldwide hire freelance scientists and industry experts on demand. Our freelancers have helped companies publish research papers, develop products, analyze data, and more. It only takes a minute to tell us what you need done and get quotes from experts for free.

Unlock Corporate Benefits
• Secure Payment Assistance
• Onboarding Support
• Dedicated Account Manager
Sign up with your professional email to avail special advances offered against purchase orders, seamless multi-channel payments, and extended support for agreements.


