



El Dr. Javier Quilez Oliete, un experimentado freelance bioinformatics consultant on Kolabtree, provides a comprehensive guide to DNA sequencing análisis de datos, including tools and software used to read data.
Introducción
El ácido desoxirribonucleico (ADN) es la molécula que transporta la mayor parte de la información genética de un organismo. (En algunos tipos de virus, la información genética es transportada por el ácido ribonucleico (ARN)). Los nucleótidos (representados convencionalmente por las letras A, C, G o T) son las unidades básicas de las moléculas de ADN. Conceptualmente, Secuenciación del ADN es el proceso de lectura de los nucleótidos que componen una molécula de ADN (por ejemplo, "GCAAACCAAT" es una cadena de ADN de 10 nucleótidos). Las tecnologías de secuenciación actuales producen millones de estas lecturas de ADN en un tiempo razonable y a un coste relativamente bajo. Como referencia, el coste de secuenciar un genoma humano -un genoma es el conjunto completo de moléculas de ADN de un organismo- ha bajado el Barrera $100 y puede hacerse en cuestión de días. Esto contrasta con la primera iniciativa de secuenciar el genoma humanoque se completó en una década y tuvo un coste de unos $2,7 mil millones.
This capability to sequence DNA at high throughput and low cost has enabled the development of a growing number of sequencing-based methods and applications. For example, sequencing entire genomes or their protein-coding regions (two approaches known respectively as whole genome and exome sequencing) in disease and healthy individuals can hint to disease-causing DNA alterations. Also, the sequencing of the RNA that is transcribed from DNA—a technique known as RNA-sequencing—is used to quantify gene activity and how this changes in different conditions (e.g. untreated versus treatment). On the other side, chromosome conformation capture sequencing methods detect interactions between nearby DNA molecules and thus help to determine the spatial distribution of chromosomes within the cell.
Estas y otras aplicaciones de la secuenciación del ADN tienen en común la generación de conjuntos de datos del orden de los gigabytes y que comprenden millones de secuencias de lectura. Por lo tanto, para dar sentido a los experimentos de secuenciación de alto rendimiento (HTS) se necesitan importantes capacidades de análisis de datos. Afortunadamente, existen herramientas computacionales y estadísticas específicas y flujos de trabajo de análisis relativamente estándar para la mayoría de los tipos de datos de HTS. Aunque algunos de los pasos (iniciales) del análisis son comunes a la mayoría de los tipos de datos de secuenciación, los análisis posteriores dependerán del tipo de datos y/o del objetivo final del análisis. A continuación, ofrezco una introducción a los pasos fundamentales del análisis de los datos de HTS y hago referencia a herramientas populares.
Algunas de las secciones siguientes se centran en el análisis de los datos generados por las tecnologías de secuenciación de lectura corta (en su mayoría Illumina), ya que éstas han dominado históricamente el mercado de HTS. Sin embargo, las nuevas tecnologías que generan lecturas más largas (por ejemplo Oxford Nanopore Technologies, PacBio) están ganando terreno rápidamente. Dado que la secuenciación de lectura larga tiene algunas particularidades (por ejemplo, mayores tasas de error), se están desarrollando herramientas específicas para el análisis de este tipo de datos.
Control de calidad (QC) de las lecturas en bruto
El analista ansioso iniciará el análisis a partir de los archivos FASTQ; el Formato FASTQ ha sido durante mucho tiempo el estándar para almacenar datos de secuenciación de hilos cortos. En esencia, los archivos FASTQ contienen la secuencia de nucleótidos y las bases por calidad de llamada para millones de lecturas. Aunque el tamaño del archivo dependerá del número real de lecturas, los archivos FASTQ suelen ser grandes (del orden de megabytes y gigabytes) y estar comprimidos. Cabe destacar que la mayoría de las herramientas que utilizan archivos FASTQ como entrada pueden manejarlos en formato comprimido, por lo que, para ahorrar espacio en el disco, se recomienda no descomprimirlos. Como convención, aquí equipararé un archivo FASTQ a una muestra de secuenciación.
FastQC es probablemente la herramienta más popular para llevar a cabo el control de calidad de las lecturas en bruto. Puede ejecutarse a través de una interfaz visual o mediante programación. Mientras que la primera opción puede ser más conveniente para los usuarios que no se sienten cómodos con el entorno de línea de comandos, la segunda ofrece una escalabilidad y reproducibilidad incomparables (piense en lo tedioso y propenso a errores que puede ser ejecutar manualmente la herramienta para decenas de archivos). En cualquier caso, el resultado principal de FastQC es un Archivo HTML reporting key summary estadísticas about the overall quality of the raw sequencing reads from a given sample. Inspecting tens of FastQC reports one by one is tedious and it complicates the comparison across samples. Therefore, you may want to use MultiQCque agrega los informes HTML de FastQC (así como de otras herramientas utilizadas posteriormente, por ejemplo, el recorte de adaptadores, la alineación) en un único informe.
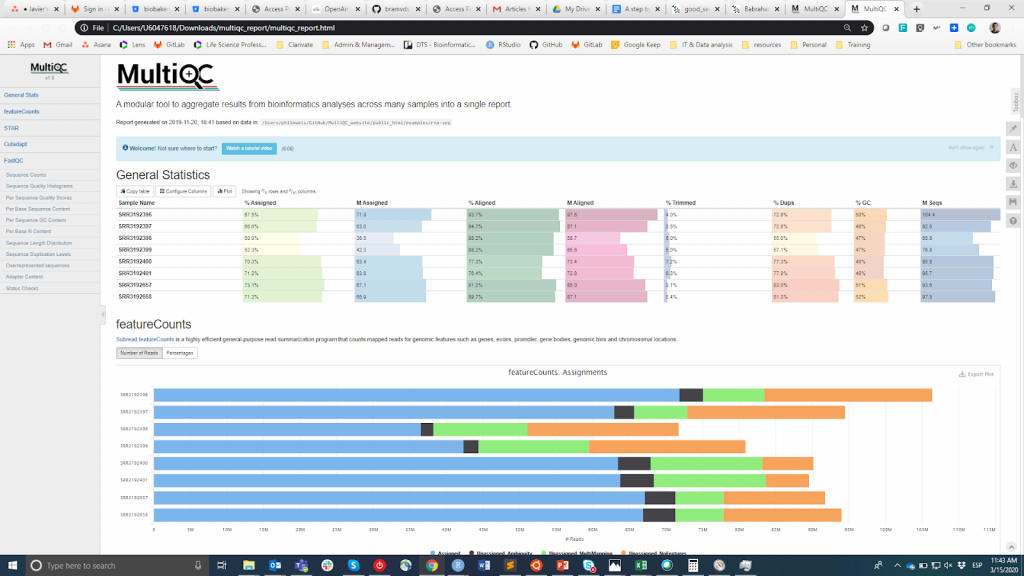
MultiQC
La información sobre el control de calidad pretende permitir al usuario juzgar si las muestras son de buena calidad y, por lo tanto, pueden utilizarse para los pasos posteriores o deben descartarse. Lamentablemente, no existe un umbral consensuado basado en la métrica FastQC para clasificar las muestras como de buena o mala calidad. El enfoque que utilizo es el siguiente. Espero que todas las muestras que han pasado por el mismo procedimiento (por ejemplo, la extracción de ADN, la preparación de la biblioteca) tengan estadísticas de calidad similares y una mayoría de indicadores de "aprobado". Si algunas muestras tienen una calidad inferior a la media, las utilizaré de todos modos en el análisis posterior teniendo esto en cuenta. Por otro lado, si todas las muestras del experimento obtienen sistemáticamente banderas de "advertencia" o "fallo" en múltiples métricas (véase este ejemplo), sospecho que algo salió mal en el experimento (por ejemplo, mala calidad del ADN, preparación de la biblioteca, etc.) y recomiendo repetirlo.
Leer el recorte
El control de calidad de las lecturas en bruto ayuda a identificar las muestras problemáticas, pero no mejora la calidad real de las lecturas. Para ello, es necesario recortar las lecturas para eliminar las secuencias técnicas y los extremos de baja calidad.
Las secuencias técnicas son restos del procedimiento experimental (por ejemplo, adaptadores de secuenciación). Si estas secuencias son adyacentes a la verdadera secuencia de la lectura, el alineamiento (véase más adelante) puede asignar las lecturas a una posición incorrecta en el genoma o disminuir la confianza en un alineamiento determinado. Además de las secuencias técnicas, también podemos querer eliminar secuencias de origen biológico si éstas están muy presentes entre las lecturas. Por ejemplo, los procedimientos de preparación del ADN subóptimos pueden dejar una alta proporción de ARN ribosómico (ARNr) convertido en ADN en la muestra. A menos que este tipo de ácido nucleico sea el objetivo del experimento de secuenciación, mantener las lecturas derivadas del ARNr sólo aumentará la carga computacional de los pasos posteriores y puede confundir los resultados. Cabe destacar que si los niveles de secuencias técnicas, ARNr u otros contaminantes son muy elevados, lo que probablemente ya habrá sido puesto de manifiesto por el control de calidad, es posible que desee descartar toda la muestra de secuenciación.
En la secuenciación de lectura corta, la secuencia de ADN se determina un nucleótido cada vez (técnicamente, un nucleótido cada ciclo de secuenciación). En otras palabras, el número de ciclos de secuenciación determina la longitud de la lectura. Un problema conocido de los métodos de secuenciación HTS es la disminución de la precisión con la que se determinan los nucleótidos a medida que se acumulan los ciclos de secuenciación. Esto se refleja en una disminución general de la calidad de la llamada por base, especialmente hacia el final de la lectura. Al igual que ocurre con las secuencias técnicas, tratar de alinear lecturas que contienen extremos de baja calidad puede llevar a una colocación errónea o a una mala calidad de mapeo.
Para eliminar las secuencias técnicas/contaminantes y los extremos de baja calidad, lea las herramientas de recorte como Trimmomatic y Cutadapt existen y se utilizan ampliamente. Básicamente, estas herramientas eliminan las secuencias técnicas (disponibles internamente y/o proporcionadas por el usuario) y recortan las lecturas en función de su calidad, al tiempo que maximizan su longitud. Las lecturas que quedan demasiado cortas después del recorte se descartan (las lecturas excesivamente cortas, por ejemplo, <36 nucleótidos, complican el paso de alineación, ya que es probable que se asignen a múltiples sitios en el genoma). Es posible que desee observar el porcentaje de lecturas que sobreviven al recorte, ya que un alto índice de lecturas descartadas es probablemente un signo de datos de mala calidad.
Por último, suelo volver a ejecutar FastQC en las lecturas recortadas para comprobar que este paso ha sido eficaz y ha mejorado sistemáticamente las métricas de control de calidad.
Alineación
Salvo excepciones (por ejemplo ensamblaje de novo), la alineación (también denominada mapeo) suele ser el siguiente paso para la mayoría de los tipos de datos y aplicaciones de HTS. La alineación de las lecturas consiste en determinar la posición en el genoma de la que se deriva la secuencia de la lectura (normalmente expresada como cromosoma:extremo inicial). Por lo tanto, en este paso se requiere el uso de una secuencia de referencia para alinear/mapear las lecturas.
La elección de la secuencia de referencia vendrá determinada por múltiples factores. Por un lado, la especie de la que procede el ADN secuenciado. Aunque el número de especies con una secuencia de referencia de alta calidad disponible está aumentando, puede que todavía no sea el caso de algunos organismos menos estudiados. En esos casos, es posible que desee alinear las lecturas con una especie evolutivamente cercana para la que exista un genoma de referencia. Por ejemplo, como no existe una secuencia de referencia para el genoma del coyote, podemos utilizar la del perro, estrechamente relacionado, para la alineación de las lecturas. Del mismo modo, es posible que queramos alinear nuestras lecturas con una especie estrechamente relacionada para la que existe una secuencia de referencia de mayor calidad. Por ejemplo, aunque el genoma del gibón ha sido publicadoEn el caso de la secuencia humana, ésta se divide en miles de fragmentos que no recapitulan totalmente la organización de ese genoma en decenas de cromosomas; en ese caso, puede ser beneficioso realizar el alineamiento utilizando la secuencia de referencia humana.
Otro factor a tener en cuenta es la versión del ensamblaje de la secuencia de referencia, ya que se publican nuevas versiones a medida que se actualiza y mejora la secuencia. Es importante destacar que las coordenadas de un determinado alineamiento pueden variar entre versiones. Por ejemplo, se pueden encontrar múltiples versiones del genoma humano en el Navegador del genoma de la UCSC. En cualquier caso, estoy totalmente a favor de migrar a la versión de ensamblaje más reciente una vez que se haya publicado por completo. Esto puede causar algunas molestias durante la transición, ya que los resultados ya existentes serán relativos a las versiones anteriores, pero vale la pena a largo plazo.
Además, el tipo de datos de secuenciación también es importante. Las lecturas generadas por los protocolos DNA-seq, ChIP-seq o Hi-C se alinearán con la secuencia de referencia del genoma. Por otro lado, como el ARN transcrito a partir del ADN se procesa posteriormente en ARNm (es decir, se eliminan los intrones), muchas lecturas de ARN-seq no se alinearán con una secuencia de referencia del genoma. En su lugar, tenemos que alinearlas con las secuencias de referencia del transcriptoma o utilizar alineadores que tengan en cuenta la división (véase más adelante) cuando se utiliza la secuencia del genoma como referencia. Relacionado con esto está la elección de la fuente para la anotación de la secuencia de referencia, es decir, la base de datos con las coordenadas de los genes, transcritos, centrómeros, etc. Yo suelo utilizar la base de datos Anotación de GENCODE ya que combina una exhaustiva anotación de genes y secuencias de transcripción.
Se ha desarrollado una larga lista de herramientas de alineación de secuencias de lectura corta (véase la sección de alineación de secuencias de lectura corta aquí). Reviewing them is beyond the scope of this article (details about the algorithms behind these tools can be found aquí). Según mi experiencia, entre los más populares están Pajarita2, BWA, HISAT2, Mapa mínimo2, STAR y TopHat. Mi recomendación es que elija su alineador teniendo en cuenta factores clave como el tipo de datos HTS y aplicación, así como la aceptación por parte de la comunidad, la calidad de la documentación y el número de usuarios. Por ejemplo, se necesitan alineadores como STAR o Bowtie2 que tengan en cuenta las uniones exón-exón al mapear el ARN-seq con el genoma.
La mayoría de los mapeadores tienen la necesidad de indexar la secuencia utilizada como referencia antes de realizar el alineamiento. Este paso puede llevar mucho tiempo, pero sólo hay que hacerlo una vez para cada secuencia de referencia. La mayoría de los mapeadores almacenan los alineamientos en archivos SAM/BAM, que siguen el esquema Formato SAM/BAM (Los archivos BAM son versiones binarias de los archivos SAM). El alineamiento es uno de los pasos que más computaciones y tiempo consume en el análisis de los datos de secuenciación y los archivos SAM/BAM son pesados (del orden de los gigabytes). Por lo tanto, es importante asegurarse de que se dispone de los recursos necesarios (véase la sección final más abajo) para ejecutar el alineamiento en un tiempo razonable y almacenar los resultados. Asimismo, debido al tamaño y al formato binario de los archivos BAM, evite abrirlos con editores de texto; en su lugar, utilice comandos de Unix o herramientas dedicadas como SAMtools.
De las alineaciones
Yo diría que no hay un paso común claro después de la alineación, ya que en este punto es donde cada tipo de datos HTS y aplicación puede diferir.
Un análisis posterior común para los datos de DNA-seq es la llamada de variantes, es decir, la identificación de posiciones en el genoma que varían en relación con la referencia del genoma y entre individuos. Un marco de análisis popular para esta aplicación es GATK para polimorfismo de un solo nucleótido (SNP) o pequeñas inserciones/deleciones (indels) (Figura 2). Las variantes que comprenden trozos más grandes de ADN (también denominadas variantes estructurales) requieren métodos de llamada específicos (véase este artículo para una comparación exhaustiva). Al igual que con los alineadores, aconsejo seleccionar la herramienta adecuada teniendo en cuenta factores clave como el tipo de variantes (SNP, indel o variantes estructurales), la aceptación por parte de la comunidad, la calidad de la documentación y el número de usuarios.
Probablemente, la aplicación más frecuente de RNA-seq es la cuantificación de la expresión génica. Históricamente, las lecturas debían alinearse con la secuencia de referencia y, a continuación, el número de lecturas alineadas con un determinado gen o transcrito se utilizaba como indicador para cuantificar sus niveles de expresión. Este enfoque de alineación+cuantificación se realiza con herramientas como Gemelos, RSEM o featureCounts. Sin embargo, este enfoque ha sido superado cada vez más por nuevos métodos implementados en software como Kallisto y Salmón. Conceptualmente, con estas herramientas no es necesario alinear la secuencia completa de una lectura con la secuencia de referencia. En su lugar, sólo necesitamos alinear suficientes nucleótidos para estar seguros de que una lectura se originó a partir de un determinado transcrito. En pocas palabras, el enfoque de alineación+cuantificación se reduce a un solo paso. Este enfoque se conoce como pseudo-mapeo y aumenta en gran medida la velocidad de la cuantificación de la expresión génica. Por otro lado, hay que tener en cuenta que el pseudo-mapeo no es adecuado para aplicaciones en las que se necesita la alineación completa (por ejemplo, la llamada de variantes a partir de datos de RNA-seq).
Otro ejemplo de las diferencias en los pasos de análisis posteriores y las herramientas necesarias en las aplicaciones basadas en la secuenciación es ChIP-seq. Las lecturas generadas con esta técnica se utilizarán para la llamada de picos, que consiste en detectar regiones en el genoma con un exceso significativo de lecturas que indica dónde se une la proteína objetivo. Existen varios "peak callers" y esta publicación los estudia. Como último ejemplo mencionaré los datos Hi-C, en los que los alineamientos se utilizan como entrada para herramientas que determinan las matrices de interacción y, a partir de ellas, las características 3D del genoma. Comentar todos los ensayos basados en la secuenciación va más allá del alcance de este artículo (para una lista relativamente completa, véase este artículo).
Antes de empezar...
La parte restante de este artículo aborda aspectos que pueden no considerarse estrictamente como pasos en el análisis de los datos de HTS y que se ignoran en gran medida. Por el contrario, sostengo que es capital que se piense en las cuestiones planteadas en Tabla 1 antes de empezar a analizar los datos de HTS (o cualquier tipo de datos, de hecho), y he escrito sobre estos temas aquí y aquí.
Tabla 1
| Piensa en ello | Acción propuesta |
| ¿Tiene toda la información de su muestra necesaria para el análisis? | Recoger sistemáticamente los metadatos de los experimentos |
| ¿Será capaz de identificar inequívocamente su muestra? | Establecer un sistema para asignar a cada muestra un identificador único |
| ¿Dónde estarán los datos y los resultados? | Organización estructurada y jerárquica de los datos |
| ¿Podrá procesar varias muestras sin problemas? | Escalabilidad, paralelización, configuración automática y modularidad del código |
| ¿Podrá usted o alguien más reproducir los resultados? | Documente su código y sus procedimientos. |
Como ya se ha mencionado, los datos brutos de HTS y algunos de los archivos generados durante su análisis son del orden de gigabytes, por lo que no es excepcional que un proyecto que incluya decenas de muestras requiera terabytes de almacenamiento. Además, algunos pasos del análisis de los datos HTS son intensivos desde el punto de vista informático (por ejemplo, la alineación). Sin embargo, la infraestructura de almacenamiento y computación necesaria para analizar los datos de HTS es una consideración importante y a menudo se pasa por alto o no se discute. Como ejemplo, como parte de un análisis reciente, revisamos decenas de artículos publicados que realizaban análisis de asociación de todo el fenotipo (PheWAS). Los PheWAS modernos analizan entre 100 y 1.000 variantes genéticas y fenotipos, lo que supone una gran capacidad de almacenamiento de datos y de computación. Y, sin embargo, prácticamente ninguno de los artículos que revisamos comentaba la infraestructura necesaria para el análisis PheWAS. No es de extrañar que mi recomendación sea que planifiquen por adelantado los requisitos de almacenamiento y computación a los que se enfrentarán y los compartan con la comunidad.
¿Necesita ayuda para analizar los datos de secuenciación de ADN? Póngase en contacto con freelance bioinformatics specialist y expertos en genómica en Kolabtree.
Kolabtree helps businesses worldwide hire freelance scientists and industry experts on demand. Our freelancers have helped companies publish research papers, develop products, analyze data, and more. It only takes a minute to tell us what you need done and get quotes from experts for free.

Unlock Corporate Benefits
• Secure Payment Assistance
• Onboarding Support
• Dedicated Account Manager
Sign up with your professional email to avail special advances offered against purchase orders, seamless multi-channel payments, and extended support for agreements.


