



Shrinidh Joshi, Medizinprodukte-Experte on Kolabtree, provides a comprehensive guide to Medizinprodukt design, design controls, validation & verification, regulatory requirements and risk management.
In der vorheriger Artikel, we took a look at the overview of the Medizinprodukt development process from the ideation to the discovery phase. In this article, we will focus more on medical device design, design controls, and compliance.
Entwurf medizinischer Geräte: IEC- und ISO-Vorschriften und deren Einhaltung
Inzwischen wissen Sie, dass Ihr Medizinprodukt für den Markteintritt bestimmte regulatorische Anforderungen und Normen erfüllen muss. Normen für Medizinprodukte wie die Internationale Elektrotechnische Kommission (IEC) oder die Internationale Organisation für Normung (ISO) ermöglichen es Herstellern von Medizinprodukten, Konstrukteuren, Labors und allen anderen Dienstleistern für die Entwicklung von Medizinprodukten, wie z. B. CDMO, ihre Geräte und Ausrüstungen nach bestimmten Qualitäts- und Gebrauchsnormen zu prüfen, zu bewerten und zu warten.
Die IEC veröffentlichte 1970 die erste Norm ihrer Art für Medizinprodukte, IEC 60601-1. IEC 60601-1, Medizinische elektrische Geräte - Teil 1: Dies ist die international anerkannte Norm, die sich mit den allgemeinen Anforderungen an medizinische elektrische Geräte und Anlagen befasst und Standards für die grundlegende Sicherheit und die wesentlichen Leistungsmerkmale enthält [4].
Das Dokument IEC 60601-1 wurde in regelmäßigen Abständen überarbeitet, um den neuesten medizinischen Entwicklungen und technologischen Durchbrüchen im Bereich der Medizinprodukte Rechnung zu tragen. Die letzte Änderung wurde 2012 vorgenommen (Änderung 1 zu IEC 60601-1). Diese überarbeitete Norm enthält die Anforderungen an die Berücksichtigung des menschlichen Faktors, die wesentliche Leistungsbewertung von Medizinprodukten, die Benutzerfreundlichkeit und die Befehle. Sie schließt auch Software als Medizinprodukt ein und legt die Einführung eines formalen Entwicklungslebenszyklus fest. Zum Geltungsbereich der überarbeiteten IEC 60601-1 gehören auch neuere und überarbeitete technische Spezifikationen für Gefahren (sowohl elektrische als auch mechanische), Anforderungen an die Kennzeichnung von Medizinprodukten (einschließlich neuer Kennzeichnungsnormen) und die Dokumentation.
Design medizinischer Geräte: ISO-Normen
Die Internationale Organisation für Normung verfügt ebenfalls über Spezifikationen für Medizinproduktnormen. ISO 13485 und ISO 14971 sind weltweit weit verbreitete Normen für das Qualitätsmanagement von Medizinprodukten. Neben diesen internationalen Normen gibt es auch einige regionsspezifische Normen, die alle mit geringen Änderungen und Einschränkungen von internationalen Normen übernommen wurden.
Wenn ein Unternehmen, das medizinische Geräte herstellt oder verkauft, in den USA tätig ist, unterliegt das medizinische Gerät der Aufsicht der FDA. Das American National Standards Institute (ANSI) ist der Vertreter der ISO-Normen in den USA.
Es gibt zwei weitere ähnliche Organisationen: die Association for the Advancement of Medical Instrumentation (AAMI) und die American Society for Quality (ASQ), die Normen für die USA definiert.
Wenn ein Medizinproduktehersteller ein Produkt unter Berücksichtigung der ISO-Normen entwickelt hat, besteht auch die Möglichkeit, dass die FDA das Produkt nicht genehmigt. Die FDA verfügt über eigene Verfahren für das Risikomanagement, die sich aus internationalen und regionalen Normen ableiten und Folgendes umfassen:
- ISO 14971:2007, Medical devices – Application of risk management to medical devices
(internationaler Standard.)
- ANSI/AAMI/ISO 14971:2007 (R2010), Medizinprodukte - Anwendung des Risikomanagements auf Medizinprodukte (Eine regionale Norm mit Ergänzungen und Änderungen gegenüber der zitierten internationalen Norm). [5].
Was die Qualitätsmanagementnorm betrifft, so folgt sie nicht der internationalen oder regionalen Version der Norm ISO 13485. Der Grund dafür ist, dass die FDA für den US-Markt andere Richtlinien für das Qualitätsmanagement von Medizinprodukten hat.
Wenn das Medizinprodukteunternehmen die Europäische Union in Betracht zieht, ist das Europäische Komitee für Normung (CEN) die von der ISO übernommene Normung und das Europäische Komitee für elektrotechnische Normung (CENELEC) die von der IEC inspirierte regionale Norm.
CEN ist entsprechend den Anforderungen der ISO etwas abgewandelt und wird mit einem "EN"-Präfix geschrieben. Z.B.:
- EN ISO 13485:2012, Medizinprodukte - Qualitätsmanagementsysteme - Anforderungen für regulatorische Zwecke.
- EN ISO 14971:2012, Medizinprodukte - Anwendung des Risikomanagements auf Medizinprodukte
Die nationalen Mitglieder übernehmen diese Normen von der EU und fügen ihr Präfix hinzu. Für die Schweiz veröffentlicht Swiss Standards Normen mit dem Präfix "SN", z. B. SN EN ISO 13485:2012 und SN EN ISO 14971:2012.
Im Falle Kanadas ist die Canadian Standards Authority (CSA) die repräsentative Organisation für die ISO.
Vorschriften für Medizinprodukte und Designkontrolle
Hersteller von Medizinprodukten müssen die Richtlinien zur Designkontrolle befolgen, da die Aufsichtsbehörden wie die FDA, die Europäische Kommission, Health Canada und andere sicherstellen wollen, dass die Medizinprodukte für potenzielle Benutzer sicher sind, bevor die Hersteller mit der Vermarktung der Geräte beginnen. Wie ich bereits im obigen Abschnitt erwähnt habe, folgt die FDA nicht der ISO 13485, da sie andere Anforderungen an das Qualitätsmanagement stellt. Designkontrollen sind definiert unter FDA 21 CFR 820.30 die einen ähnlichen Zweck verfolgt wie Abschnitt 7.3 Design und Entwicklung, der in den Leitlinien für ISO 13485 beschrieben ist. Darüber hinaus hat die FDA die Anforderungen der "Current Good Manufacturing Practice" (cGMP) in die Vorschriften für das Qualitätssystem aufgenommen, um gute Qualitätspraktiken für die Entwicklung von Medizinprodukten einzuhalten [6].
Die Verordnung bietet einen Rahmen für die Umsetzung der Entwurfskontrolle bei einer Vielzahl von Geräten. Der Rahmen bietet Flexibilität sowohl für die Einhaltung von Vorschriften als auch für den internen Entwurfs- und Entwicklungsprozess.
Für die erfolgreiche Umsetzung der Entwurfskontrolle von Medizinprodukten werden Fachleute mit technischem und nichttechnischem Hintergrund benötigt, z. B. aus den Bereichen Betriebswirtschaft, Biowissenschaften, Ingenieurwesen, Informatik und Kunst.
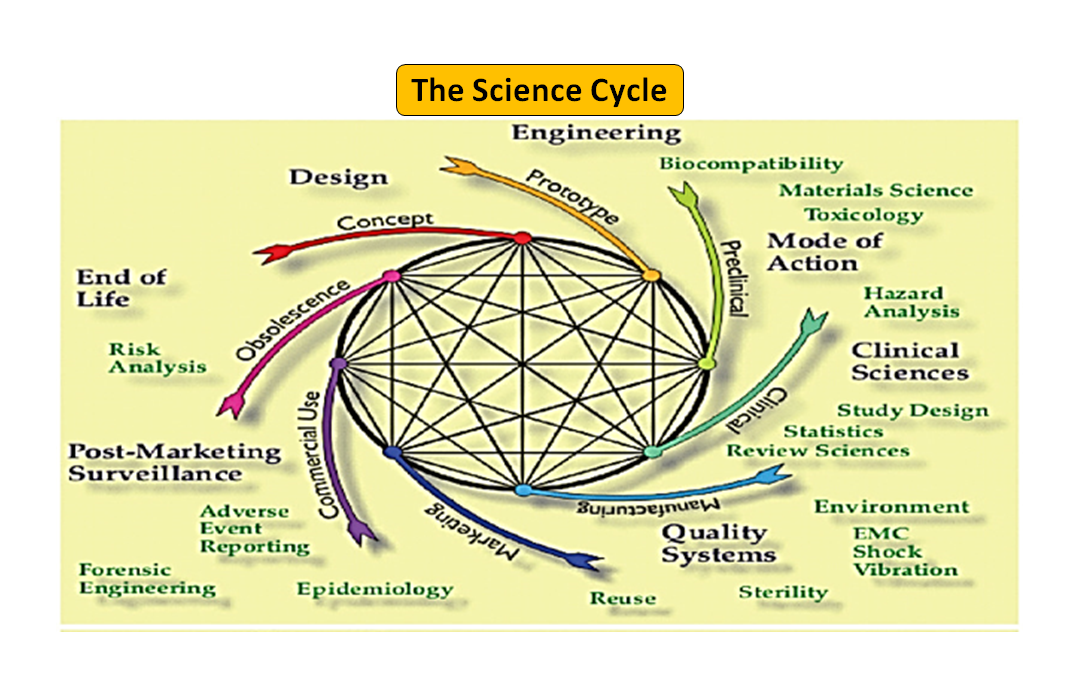
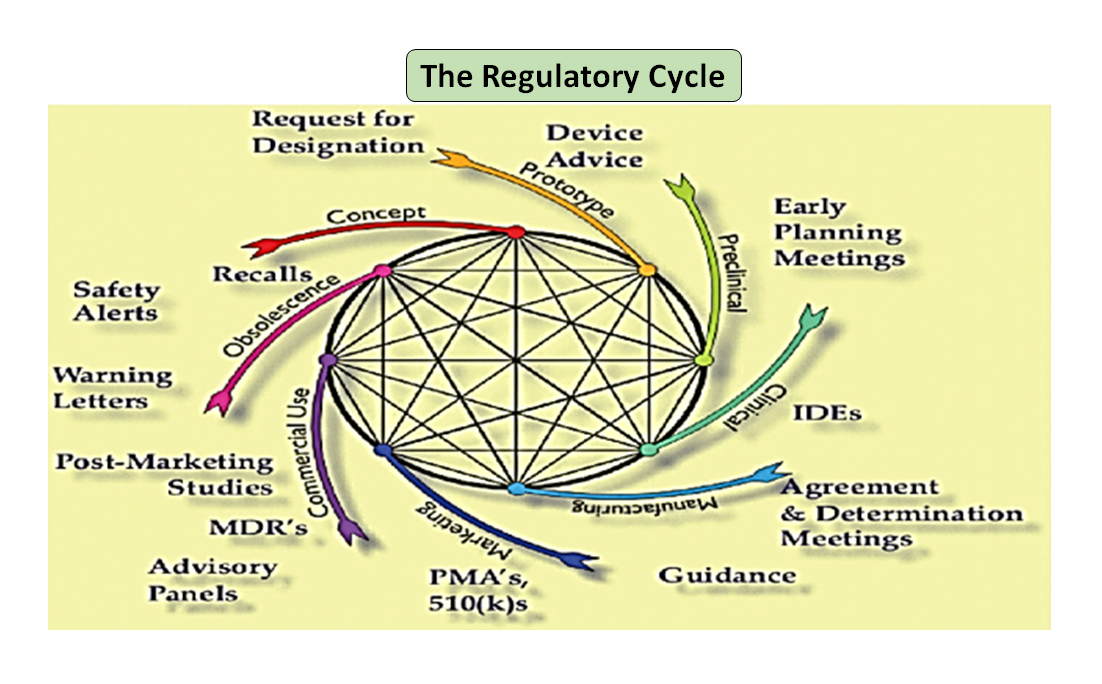
Abbildung 1: Der gesamte Produktlebenszyklus. Der Wissenschaftszyklus und der Regulierungszyklus (Nach einer Vorlage von [7]).
Es ist bemerkenswert, dass der Lebenszyklus eines Medizinprodukts von der Innovation bis zur behördlichen Zulassung und Kommerzialisierung eine Reihe von miteinander verbundenen Schritten ist, die die Produktentwicklung vorantreiben (siehe Abbildung 1: Gesamter Produktzyklus). Zu Beginn werden die von Ihren Ingenieuren entworfenen Prototypen auf dem Prüfstand getestet, um das Design zu optimieren, und auf Biokompatibilität, extrahierbare und auslaugbare Bestandteile, Flexibilität und allgemeine Festigkeit Ihres Produkts geprüft. Die Aufgabe des Regulierungsberaters Ihres Unternehmens besteht darin, die Datenbank der Regulierungsbehörden zu durchsuchen und Ihnen einen Leitfaden vorzuschlagen, mit dessen Hilfe Sie feststellen können, ob Ihr Produkt als Medizinprodukt reguliert werden kann oder nicht. Die vorgesehene Verwendung Ihres Medizinprodukts und seine Funktions- oder Wirkungsweise dienen als Leitfaden für die Gestaltung des Produkts und entscheiden auch über den Zulassungsweg, ob 510(k), PMA, De Novo, Pre-sub, IDE, HDE, Master Files usw.
Wie in Abbildung 1 dargestellt, sind sowohl die wissenschaftlichen als auch die regulatorischen Prozesse während des gesamten Produktlebenszyklus miteinander verflochten. Genauso wie die verschiedenen Teile des wissenschaftlichen Lebenszyklus miteinander verbunden sind, sind auch die wissenschaftlichen und regulatorischen Anforderungen miteinander verflochten, wobei die eine die andere informiert und bestimmt. Es besteht die Möglichkeit, sowohl bei der FDA als auch bei den Herstellern Verbindungen herzustellen, damit Teile des Lebenszyklus nicht Gefahr laufen, nur isoliert betrachtet zu werden. So ist es zum Beispiel nicht ungewöhnlich, dass ein Antrag vor dem Inverkehrbringen geprüft wird, ohne die Erfahrungen mit ähnlichen Produkten nach dem Inverkehrbringen zu berücksichtigen.
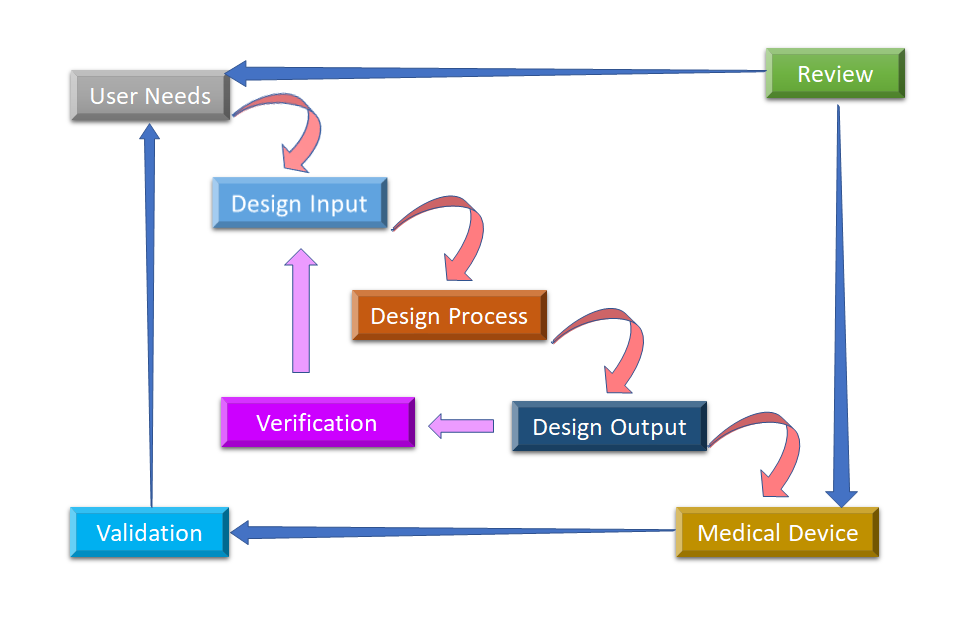
Abbildung 2: Wasserfluss-Designprozess für die Designkontrolle von Medizinprodukten (angepasst von [8]).
Die erste Phase, mit der die Designkontrolle beginnt, ist die Entwicklung und Genehmigung des Design-Inputs, der aus dem Gerätedesign und den in der Produktionsphase durchzuführenden Herstellungsprozessen besteht. Die Designkontrolle ist ein ganzheitlicher Ansatz und endet nicht mit der Übertragung des Designs in die Produktionsphase, sobald das Design fertiggestellt ist. Sie wirkt sich auch auf die Herstellungsprozesse entsprechend den Änderungen in der Entwurfsphase oder sogar auf das Feedback nach der Produktion aus. Es ist ein fortlaufender Prozess, ein Produkt zu entwickeln, das für den Benutzer brauchbar ist, und daher werden bei der Produktverbesserung revolutionäre Veränderungen aufgrund von Nutzungsmustern ebenso berücksichtigt wie die Analyse misslungener Produkte. In Abbildung 2 sehen Sie, wie die Designkontrolle im Wasserfall-Designprozess durchgeführt werden kann.
- Benutzerbedürfnisse:- Die Anforderungen werden unter Berücksichtigung der Marktbedürfnisse definiert, und das Gerät wird so konzipiert, dass es diese Bedürfnisse erfüllt. Nach einer Reihe von Entwicklungsschritten wird das Design des Medizinprodukts fertiggestellt und zur Herstellung an die Produktion übergeben. Bei jedem Schritt dieses Prozesses ist ein Feedback erforderlich.
- Entwurfseingabe: Dies ist ein iterativer Prozess. Wenn eine Organisation beschließt, sich mit einem bestimmten Bedarf zu befassen, prüft sie die Annehmbarkeit des aus dem Bedarf abgeleiteten Designinputs und testet ihn. An diesem Punkt beginnt der iterative Prozess der Umwandlung von Anforderungen in ein Gerätedesign.
- Entwurfsprozess: Diese Design-Inputs werden in Design-Output umgewandelt, indem diese Anforderungen in High-Level-Spezifikationen umgewandelt werden (die Design-Output sind).
- Entwurfsleistung: Der Verifizierungsprozess bestätigt, ob die Spezifikationen den Anforderungen genügen oder nicht. Der Output wird zum Input für die Überarbeitung der Anforderungen und dieser Prozess geht weiter, bis der Design-Output mit dem Design-Input übereinstimmt.
- Medizinisches Gerät: Once the final design is ready, it is transmitted to the production facility for mass manufacturing. Design control regulation mandates Design History File (DHF), which illustrates the linkages and relationships between all the Design Controls and help to trace all changes throughout the entire Produktentwicklung Prozess.
Medizintechnikunternehmen können einen papierbasierten oder einen softwarebasierten Ansatz wählen, der speziell für die Designkontrolle entwickelt wurde; die Dateien der Designhistorie müssen rückverfolgbar und für alle Teammitglieder zugänglich sein.
Das nachstehende Flussdiagramm zeigt eine Fallstudie zur Kontrolle der Entwicklung medizinischer Geräte.

Design medizinischer Geräte: Warum Rückverfolgbarkeit wichtig ist
Currently in the realm of the medical devices industry, it is an ideal practice to develop a traceability matrix that can illustrate the links and relations between user needs, design inputs and outputs, design verification and validation. When you are in the early phase for your device development you can maintain the device traceability using a spreadsheet or document version but as you move forward, its good idea to use cloud-based project management and document sharing platforms such as Microsoft Teams, Asana, Trello or whichever platform is suitable for your organization. The goal is as your project progresses you need to find an option which can save time because the old-school method of maintaining a traceability matrix might consume a lot of your time which you should rather be focusing on design verification and validation.
Eine Rückverfolgbarkeitsmatrix für Entwurfskontrollen ist für Produktentwicklungsteams und insbesondere für Projektmanager von entscheidender Bedeutung, da die Rückverfolgbarkeit die Beziehungen und Verknüpfungen zwischen allen Entwurfskontrollen aufzeigt. Wie verhalten sich die Benutzerbedürfnisse zu den Design-Inputs? Wie hängen die Design-Outputs mit den Design-Inputs zusammen? Wie hängen Entwurfsüberprüfungen mit Entwurfseingaben und Entwurfsausgaben zusammen? Wie beziehen sich Design-Validierungen auf die Benutzerbedürfnisse? Eine Rückverfolgbarkeitsmatrix ist ein unschätzbares Werkzeug, um eine Übersicht und den Fluss der Produktentwicklung von Medizinprodukten von Anfang bis Ende darzustellen.
Best-Practice-Produktentwickler verlassen sich schon seit vielen, vielen Jahren auf die Rückverfolgbarkeit von Design Controls. Und jetzt ISO 13485:2016 macht auch die Rückverfolgbarkeit zu einer Anforderung. Wie in ISO 13485:2016, 7.1 Planung der Produktrealisierung, 1. c) erforderliche Verifizierungs-, Validierungs-, Überwachungs-, Mess-, Inspektions- und Prüf-, Handhabungs-, Lagerungs-, Vertriebs- und Rückverfolgbarkeitsaktivitäten, die für das Produkt spezifisch sind, zusammen mit den Kriterien für die Produktakzeptanz; und 7.3.2 Entwurfs- und Entwicklungsplanung, 1. e) die Methoden zur Sicherstellung der Rückverfolgbarkeit von Entwurfs- und Entwicklungsergebnissen zu Entwurfs- und Entwicklungseingaben [9].
Entwurf medizinischer Geräte: Verifizierung und Validierung
Jedes medizinische Gerät muss die Anforderungen an Funktionalität, Benutzerfreundlichkeit und Zuverlässigkeit erfüllen, um sich auf dem Markt durchsetzen zu können.
Darüber hinaus werden Ihre Interessengruppen (Patienten, Verordner, Regulierungsbehörden oder Endnutzer) auch auf die Sicherheit und Wirksamkeit Ihres Produkts achten. Es ist sehr wahrscheinlich, dass Ihr Produkt auf einen ungedeckten Bedarf abzielt, der lebenswichtig sein kann, z. B. ein Beatmungsgerät oder ein Diagnosegerät zur Erkennung von Herzkrankheiten. Daher ist die iterative Prüfung Ihres Geräts mit Verifizierung und Validierung von entscheidender Bedeutung. Diese beiden Schritte im Entwicklungsprozess sollen bestätigen, dass Ihr medizinisches Gerät den Anforderungen der Nutzer entspricht und seine Leistung entsprechend der vorgesehenen Verwendung erbringt. Einfach ausgedrückt, können Designverifizierung und -validierung sicherstellen, dass Ihr Gerät tatsächlich das tut, was es tun soll. Designverifizierung und -validierung dienen auch dazu, die gesetzlichen Anforderungen, Normen, Produktqualität und den Herstellungsprozess Ihres Medizinprodukts sicherzustellen. Mit der Entwurfsprüfung kann bewertet werden, ob die Ergebnisse Ihres Entwurfs mit den festgelegten Anforderungen, Spezifikationen oder behördlichen Auflagen übereinstimmen, die im Entwurfsinput angegeben sind. Andererseits soll die Designvalidierung bewerten, ob Ihr Medizinprodukt den Bedürfnissen der Endnutzer entspricht.
Gestaltung Überprüfung fragt: "Haben wir das Gerät richtig entworfen?"
Gestaltung Validierung fragt: "Haben wir das richtige Gerät entworfen?"
Medizinische Geräte können verschiedene technologische Formen, Größen und unterschiedliche Komplexitätsgrade aufweisen. Die Verifizierungs- und Validierungsaktivitäten (V&V) werden durch das regulatorische Umfeld bestimmt und müssen internationalen Standards entsprechen. Standardisierte V&V-Aktivitäten können sowohl den Herstellungsprozess rationalisieren als auch das Zulassungsverfahren verbessern. Darüber hinaus können automatisierte Tests, Diagnosetechniken und Datenerfassungstools den V&V-Prozess verbessern. [10].
- Produktvalidierung vs. Prozessvalidierung
- Design von Medizinprodukten/Produktvalidierung: - Entspricht das Gerät den Bedürfnissen der Nutzer und Patienten, d. h. funktioniert es richtig?
- Prozessvalidierung:- Der Herstellungsprozess entspricht den vorgegebenen Spezifikationen.
Es ist zu beachten, dass Entwurfs-/Produktvalidierung ≠ Prozessvalidierung. Die Zulassungsbehörden verlangen sowohl die Entwurfs-/Produktvalidierung als auch die Prozessvalidierung einzeln, so dass beide bei der Einreichung von Zulassungsanträgen gleichermaßen berücksichtigt werden müssen.
Wie früh im Entwicklungsprozess sollten wir an die Validierung denken? Ein Medizintechnikunternehmen sollte verstehen, dass es nie zu früh ist, mit der Validierung zu beginnen. Ein Unternehmen sollte eher früher als später mit der Validierung beginnen, um herauszufinden, ob es den richtigen Weg einschlägt und das richtige Problem löst.
Da es sich bei der Validierung (auch V&V) um einen iterativen Prozess handelt, verschlingt sie eine gute Investition, wenn sie schlecht geplant ist. Eine klar definierte Teststrategie kann Ihnen helfen, die Kosten und die Testdauer zu optimieren, um das Produkt rechtzeitig zur Marktreife zu bringen.
Die Komplexität einer Teststrategie hängt von den zu verwendenden Technologien und den geografischen Zielmärkten ab. Die Teststrategie sollte mindestens sechs der unten genannten Parameter abdecken:
- Geografische Zielgebiete und damit verbundene Standards;
- Zeit bis zur Markteinführung;
- Ein Standard, der mit einer Version befolgt wird;
- Prüflabore - interne oder unabhängige Labore;
- Festlegung der Reihenfolge der Tests;
- Präsentation des Testergebnisses
Dementsprechend müssen auch die Tests, die für den Verifizierungs- und Validierungsprozess verwendet werden, validiert werden. Damit soll sichergestellt werden, dass Sie das messen, was Sie messen müssen, denn ein falscher Test liefert falsche Ergebnisse in Bezug auf Benutzerfreundlichkeit und Funktionalität. Medizintechnikunternehmen benötigen eine wirksame und gut dokumentierte V&V, die den einschlägigen Vorschriften entspricht.
Design medizinischer Geräte: Risikomanagement
Risikomigrationsstrategie vs. Risikomanagementplan
Die Risikomanagementverfahren für Medizinprodukte werden im Rahmen international anerkannter Konformitätsstandards durchgesetzt ISO 14971:2007 Medizinprodukte - "Anwendung des Risikomanagements auf Medizinprodukte". Abgesehen davon müssen Risikomanagementmaßnahmen in alle Phasen der Konzeption und Entwicklung von Medizinprodukten einbezogen werden und sollten auch mit Aspekten der Auslegungskontrolle verbunden sein [10].
Das Risikomanagement endet nie (zumindest theoretisch!). Die Philosophie des Risikomanagements besteht darin, dass es sich nicht um ein festes Regelwerk handeln sollte. Beim Risikomanagement und bei der Strategie der Risikomigration geht es darum, die Absicht des Risikomanagements zu verstehen und den Prozess logisch und systematisch anzugehen. Mit anderen Worten, Befolgen Sie nicht nur die Regeln... denken Sie nach!
Considering the complexity of medical device design, focused risk management practices help ensure usability, safety, and Einhaltung von Rechtsvorschriften. It is a process of identifying, controlling, and preventing the failure that may cause hazards to users. It also mandates identifying associated risks.
Abbildung 3 zeigt alle Schritte des Risikomanagementprozesses. Der Prozess beginnt mit der Identifizierung von Gefahren, und dann wird das damit verbundene Risiko auf der Grundlage der Folgen der Gefahren und ihrer möglichen Risiken gemessen.
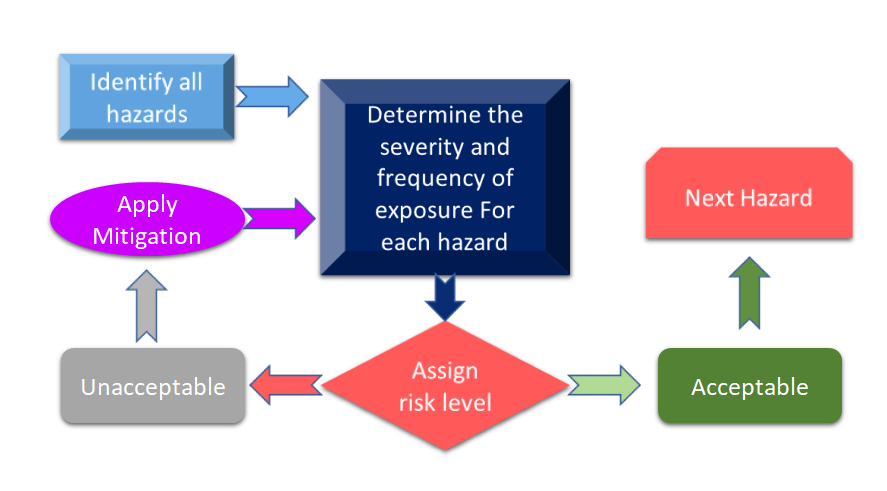
Abbildung 3: Risikomanagementprozess für Medizinprodukte (Angepasst von [11]).
Wenn das im Rahmen des Risikomanagementprozesses für Medizinprodukte ermittelte Risiko über den festgelegten Kriterien liegt, müssen Sie eine Risikominderung vornehmen. Die Höhe des Risikos hängt von verschiedenen Parametern ab, unter anderem von Ihrem Produkt, den Technologien und in einigen Fällen auch von der Art und Weise, wie Ihr Unternehmen den Prozess der Risikominderung handhabt. Es ist immer ratsam, eine Gefahrenanalyse für Ihr Gerät durchzuführen, um zu sehen, welche Normen auf Ihr Gerät angewendet werden können. In der jüngsten Überarbeitung der ISO 14971: International Standard for Risk Management of Medical Devices (Internationale Norm für das Risikomanagement von Medizinprodukten) werden die Risikoanalyse und die vorläufige Gefahrenanalyse (Preliminary Hazard Analysis - PHA) als Hauptanforderungen für Ihr Medizinprodukt genannt. [12]. Vereinfacht ausgedrückt soll die PHA den anfänglichen Rahmen für Risikobewertungen und -management bilden, und die PHA umfasst sowohl die Risikoanalyse als auch die Risikobewertung. Laut Definition umfasst die PHA eine Liste von Gefahren, Schäden und gefährlichen Situationen, die sich aus den Konstruktionsmaterialien (MoC) Ihrer Geräte, den in Ihrem Gerät verwendeten Komponenten oder Rohstoffen, den Schnittstellen zwischen Mensch und Gerät oder den manuellen Schnittstellen, der Einsatzumgebung, dem Funktionsprinzip und anderen relevanten Faktoren ergeben. [13].
Schlussfolgerung
Letztendlich ist es für jedes neu gegründete oder bereits etablierte Unternehmen im Bereich Medizinprodukte wichtig, sich daran zu erinnern, dass das Lesen der Vorschriften nichts bringt, aber das Verstehen der Philosophie sehr viel!
Fazit: Wenn es um Risikoanalyse und Planung geht:
- Sie sollten frühzeitig und während des gesamten Design- und Entwicklungsprozesses eingesetzt werden,
- Häufig werden neue Informationen generiert, die in den Design- und Entwicklungsprozess einfließen (für aktuelle und zukünftige Geräte),
- Keine noch so gute Planung kann alle Gefahren und Risiken ausschalten... aber Sie können viele von ihnen abmildern! (Die Befolgung der hier beschriebenen Entwurfskontrollphilosophien mindert das Risiko automatisch!)
Der Weg eines jeden Medizinprodukts bis zur Marktreife ist aufgrund der verschiedenen zu berücksichtigenden Faktoren wie Nutzungsmuster, Material, Benutzererfahrung, Vorschriften und mehr komplex.
Brauche Hilfe bei medical device design? Browse experienced Experten der Medizintechnikbranche auf Kolabtree oder stellen Sie Ihr Projekt kostenlos ein, um Vorschläge zu erhalten.
REFERENZEN UND RESSOURCEN
- https://www.welldoc.com/health-plans/
- https://ec.europa.eu/docsroom/documents/10337/attachments/1/translations
- FDA, 2005, Total Product Lifecycle, FDA-CDRH Präsentation von CDRH-Direktor Dr. David Feigal, http://www.fda.gov/cdrh/strategic/presentations/ tplc.html
- Pietzsch, Jan & Shluzas, Lauren & Paté-Cornell, Marie-Elisabeth & Yock, Paul & Linehan, John. (2009). Stage-Gate-Prozess für die Entwicklung von Medizinprodukten. Zeitschrift für Medizinprodukte. 3(2).
- Regulierungsstrategien für die dritte Ausgabe der IEC 60601-1, abgerufen am 9. September 2020.
- https://www.meddeviceonline.com/doc/an-introduction-to-international-medical-device-standards-0001
- https://www.fda.gov/files/drugs/published/Design-Controls—Devices.pdf
- Feigal DW. Anhang D. Auswirkungen des regulatorischen Rahmens auf die Entwicklung und Innovation von Medizinprodukten. Institut für Medizin (US) Ausschuss für die Öffentliche Gesundheit Effectiveness of the FDA 510(k) Clearance Process; Wizemann T, editor. Public Health Effectiveness of the FDA 510(k) Clearance Process: Balancing Patient Safety and Innovation: Workshop Report. Washington (DC): National Academies Press (US); 2010. Appendix D, Impact of the Regulatory Framework on Medical Device Development and Innovation. Available from: https://www.ncbi.nlm.nih.gov/books/NBK209794/.
- 1997, FDA CDRH 1997, Design Control Guidance for Medical Device Manufacturers
- https://starfishmedical.com/blog/iso-134852016-section-7/?doing_wp_cron=1599995964.4528369903564453125000
- Teixeira, M. B., und Bradley, R., 2003, Design Controls for the Medical Device Industry, Marcel Dekker, New York.
- ISO 14971:2019 - Medizinprodukte - Anwendung des Risikomanagements auf Medizinprodukte
- ISO/TR 24971:2020 - Medizinprodukte - Leitfaden für die Anwendung der ISO 14971
Alle Artikel dieser Serie:
Entwicklung und Design von Medizinprodukten: Ein definitiver Leitfaden
Entwicklung medizinischer Geräte: 3 Tipps für den Erfolg
Entwurf medizinischer Geräte: Der wesentliche, schrittweise Leitfaden
Vermarktung von Medizinprodukten: 9 Schritte von der Skizze bis zur Markteinführung
Wie man die Herausforderungen bei der Kommerzialisierung von Medizinprodukten meistert
Markteinführung medizinischer Geräte: Die wichtigsten Schritte zur Markteinführung Ihres Produkts
Post-Market Surveillance von Medizinprodukten: Ein umfassender Leitfaden
Kolabtree helps businesses worldwide hire freelance scientists and industry experts on demand. Our freelancers have helped companies publish research papers, develop products, analyze data, and more. It only takes a minute to tell us what you need done and get quotes from experts for free.

Unlock Corporate Benefits
• Secure Payment Assistance
• Onboarding Support
• Dedicated Account Manager
Sign up with your professional email to avail special advances offered against purchase orders, seamless multi-channel payments, and extended support for agreements.


