



An exhaustive guide to Einhaltung von Rechtsvorschriften for IVD manufacturers, written by Sundeep Agarwalerfahren IVDR-Berater.
Was ist die IVDR?
The European Commission’s (EC) In Vitro Diagnostic Regulation (EU IVDR 2017/746) is a ‘legislative framework’ and a way forward towards global IVD safety, which assures that only reliable and effective IVDs are in the market. The European Commission is trying its best to make the Gesundheitswesen system safer and error free in terms of diagnosis or outcomes.
Die In-vitro-Diagnostika (IVDD) war 98/79/EG eine Richtlinie, während IVDR eine Gesetzgebung (Verordnung) ist, die für alle Wirtschaftsbeteiligten (EO) gilt, d. h. für Hersteller, Importeure, Anwender, benannte Stellen und nationale Behörden im Europäischen Wirtschaftsraum (EWR) sowie für Nicht-EU-Hersteller und -Lieferanten, die IVD auf dem europäischen Markt in Verkehr bringen oder zu vertreiben planen.
Die IVDR besteht aus 113 Artikeln (10 Kapiteln) und fünfzehn Anhängen im Vergleich zu 24 Artikeln und zehn Anhängen der IVDD. Zweifelsohne ist die IVDR ein langwieriges und sehr strenges Regelwerk, aber das Gute daran ist, dass sie transparenter ist, was die Änderungen und Anforderungen betrifft.
Der Schwerpunkt liegt auf dem lebenszyklusbasierten Ansatz. Sie wird ab dem 26. Mai 2022 angewandt, und es wird erwartet, dass die Wirtschaftsakteure (einschließlich Nicht-EU-Hersteller) sich proaktiv auf die Planung und Umsetzung derselben vorbereiten. Alle am Prozess Beteiligten sind nun gleichermaßen für den In-vitro-Diagnostikmarkt im Europäischen Wirtschaftsraum (EWR) verantwortlich.
- Das erste und wichtigste, was eine Organisation tun sollte, ist, ein Schulungsprogramm (online oder vor Ort, je nachdem) zur EU IVDR zu organisieren, damit jeder in der Organisation die notwendigen Änderungen kennt.
- Es sollte eine offizielle Mitteilung an alle Lieferanten, Unterauftragnehmer oder Dienstleister über das Verfahren und ihre Pflichten erfolgen.
- Führen Sie eine Lückenbewertung durch, um die Verfügbarkeit ihrer Ressourcen und eines kompetenten Teams für die Aktualisierung der nach der EU-IVDR erforderlichen technischen Dokumentation zu prüfen. Eine Zertifizierung nach ISO 13485: 2016 wäre ein zusätzlicher Vorteil, um die Konformität herzustellen.
- Es ist ratsam (falls erforderlich), bereits in der Anfangsphase der Umstellung einen Fachexperten oder einen externen Berater hinzuzuziehen, denn "was lange währt, wird endlich gut".
- Dieser Blog bietet einen detaillierten Überblick und praktische Tipps, um die Erwartungen der benannten Stellen und zuständigen Behörden zu erfüllen, wie sie in den verschiedenen Artikeln und Anhängen der EU IVDR 2017/746 beschrieben sind.
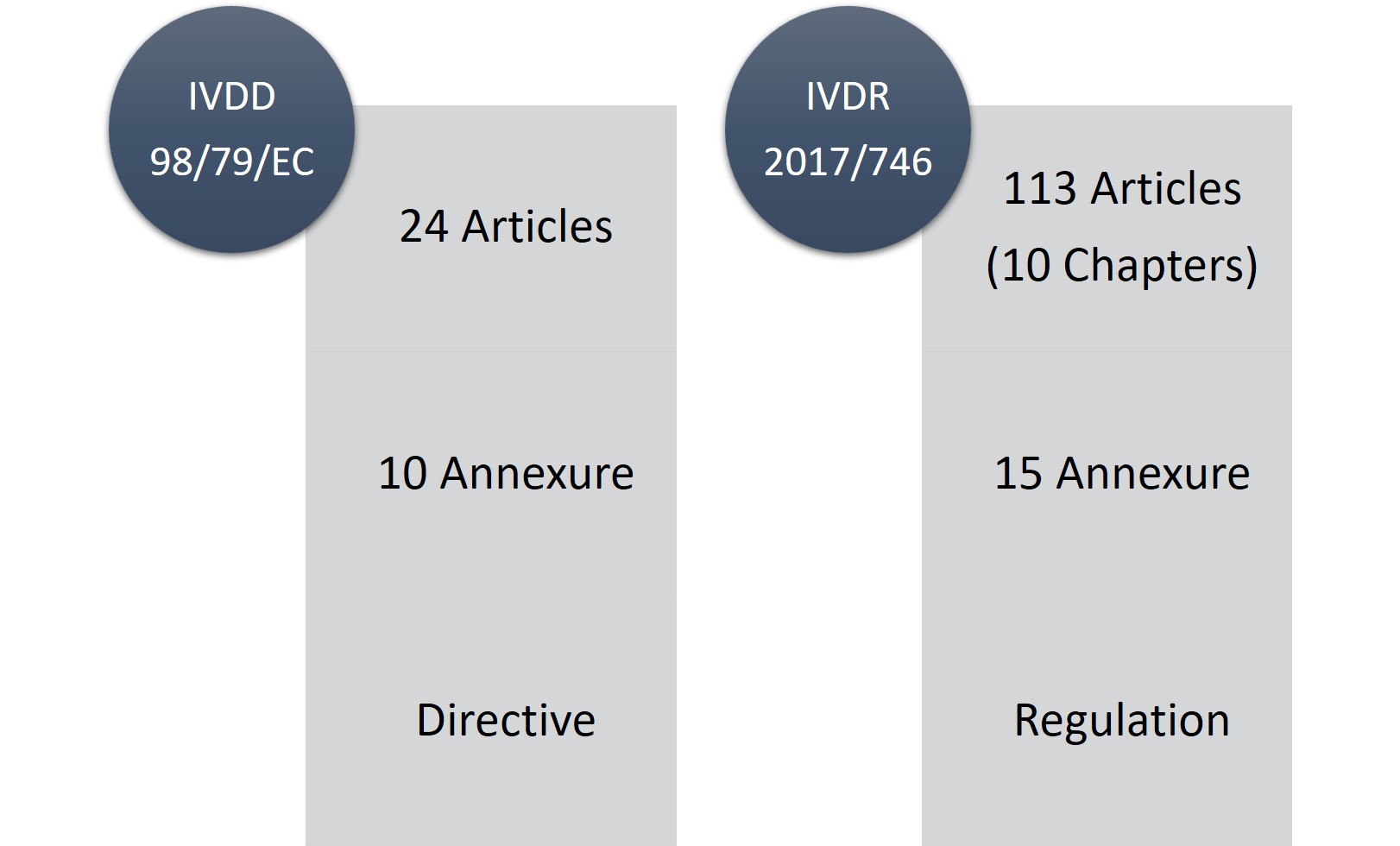
Abbildung 1: IVDD vs. IVDR
1. Vorbereitung auf die Einhaltung der IVDR und kommerzielle Änderungen
Die wichtigste geschäftliche Entscheidung für eine Organisation wäre die Entscheidung, ob sie ihre IVD weiterhin im Europäischen Wirtschaftsraum (EWR) in Verkehr bringen will. Wenn die Antwort "ja" lautet, sollte man so früh wie möglich Kostenvoranschläge (Kosten), Zeitpläne, Umfang des Audits, Produktcode usw. von einer BS einholen. Der Übergang von einer Richtlinie zu einer Verordnung erfordert eine verbindliche Einhaltung der Vorschriften und eine solide technische Dokumentation, um die Sicherheit und Wirksamkeit nachzuweisen und die CE-Zertifizierung zu erhalten. Die IVDR stützt sich weitgehend auf die klinischen Nachweise d. h. wissenschaftliche Gültigkeit, analytische Leistung und klinische Leistung um die Sicherheit und Wirksamkeit festzustellen.
Die Einbeziehung einer benannten Stelle (BS) in den Prozess der CE-Zertifizierung wird ein wichtiges Merkmal der Verordnung sein. Dies bedeutet auch eine zusätzliche Investition für den Wirtschaftsteilnehmer, die indirekt die Kosten des Produkts erhöhen kann.
Die Ernennung eines "Für die Einhaltung der Rechtsvorschriften verantwortliche Person (PRRC)" in accordance with Article 15 of EU IVDR 2017/746 is now mandatory; who shall assure the conformity of QMS, declaration of conformity, technical documentation, post market surveillance and reporting of adverse events are in compliance to EU IVDR.Manufacturers should ensure that the entire transition (including new certification application) is completed before the expiry of their existing IVDD Certificate or Self-certified Declaration of conformity. Certificates issued by notified bodies in accordance with IVDD 98/79/EC from 25 May 2017 shall become invalid after 27 May 2024. Be aware of the new timeline for application as per the EC official press release [1] dtd.20th Dezember 2022.
2. Klares Verständnis der Klassifizierung
Überprüfen Sie die neue Klassifizierungsregel gemäß Anhang VIII der IVDR und prüfen Sie, ob sie sich auf Ihre bisherige Klassifizierung ausgewirkt hat.
Eine korrekte Klassifizierung ist unerlässlich, bevor wir uns auf das CE-Zertifizierungsverfahren vorbereiten. Wenn wir dazu nicht in der Lage sind, wird der Konformitätsweg unklar sein und unsere Bemühungen um die Einhaltung der IVDR-Anforderungen verzögern oder zunichte machen.
Die IVDR ist eine risikobasierter Ansatz Einstufung der Produkte mit verstärkten Kontrollen durch die benannte Stelle und die zuständige Behörde. In der Verordnung wird Folgendes festgelegt vier Risikoklassen: Klasse A (geringstes Risiko), Klasse B, Klasse C und Klasse D (höchstes Risiko), während in Anhang VIII definiert wird sieben Klassifizierungsregeln um die Produkte korrekt zu klassifizieren. Ein einzigartiges Merkmal der IVDR ist, dass auch Software gemäß der Durchführungsvorschrift 1.4 des Anhangs VIII klassifiziert wird, in der es heißt: "Software, die ein Produkt steuert oder die Verwendung eines Produkts beeinflusst, fällt in dieselbe Klasse wie das Produkt. Wenn die Software unabhängig von einem anderen Produkt ist, wird sie als eigenständige Klasse eingestuft.[2]". Dies zeigt den Anwendungsbereich der Software an, die unter der IVDR geregelt wird. Und der Hersteller muss auch die Verifizierung und Validierung der Software (Anhang II, 6.4) entsprechend durchführen.
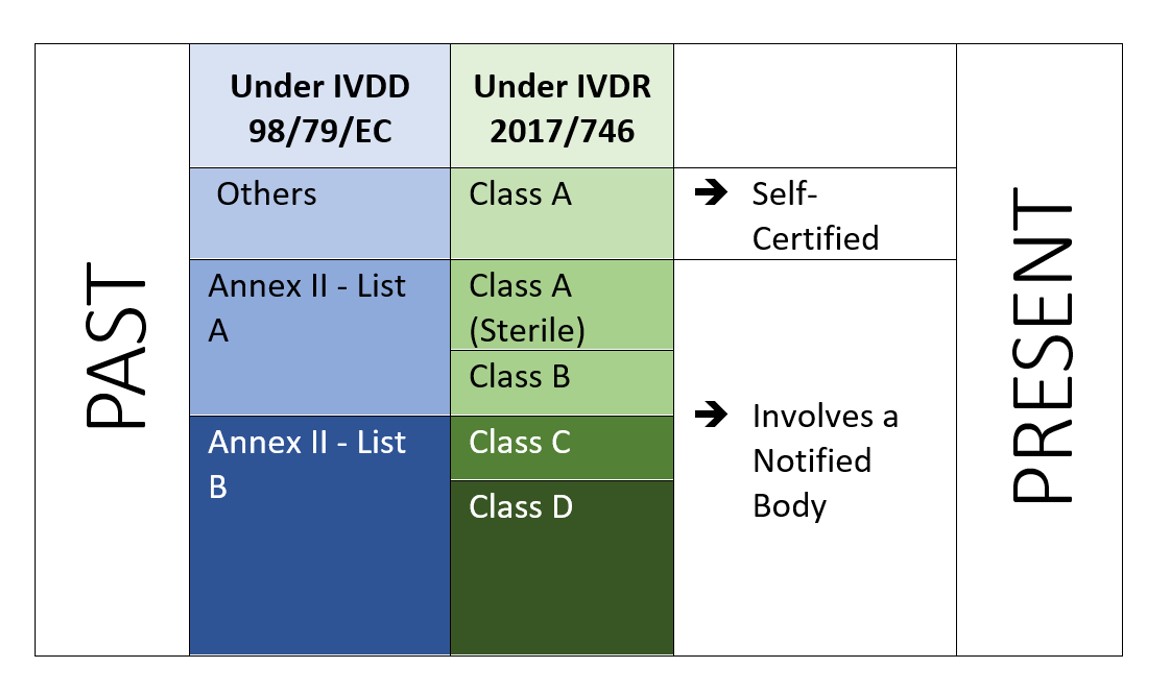
Abbildung 2: Risikobasierte Klassifizierung gemäß IVDR 2107/746
3. Einbeziehung der benannten Stelle
Die Rolle einer benannten Stelle (NB) wäre eines der Kernelemente, so dass eine größere Anzahl von Herstellern nun von einer benannten Stelle auditiert und zertifiziert werden müsste, im Gegensatz zur traditionellen Methode der "Selbstzertifizierung".Die Wirtschaftsakteure sollten sorgfältig über den Weg der Konformitätsbewertung entscheiden (Anhang IX, X, XI der EU IVDR[3]).
Die IVDR erfordert nicht nur zusätzliche Investitionen, sondern sie müssen auch sicherstellen, dass ihre technische Dokumentation und ihr Qualitätsmanagementsystem den neuen Anforderungen der IVDR entsprechen. Im Rahmen der IVDD sind die meisten IVD selbstzertifiziert (92%) und erfordern nicht die Einschaltung einer benannten Stelle (mit Ausnahme von 8% der insgesamt auf dem Markt befindlichen IVD[4]). Unter der neuen IVDR ist das Szenario jedoch anders.
Laut einer Studie “The impact of the new European IVD-classification rules on the notified body involvement” by National Institute for Öffentliche Gesundheit and the Environment, Bilthoven (Netherlands) RIVM Letter report 2018-0082, A. van Drongelen et al., nearly 85% of all IVDs will be requiring Notified Body involvement, leaving only 15% of IVDs eligible for self-certification[5].
Dies bedeutet auch, dass die Hersteller von In-vitro-Diagnostika (IVDs) nun eine große Umstellung erleben werden, um dem neuen Klassifizierungs- und Zertifizierungsverfahren zu entsprechen. Je nach Verwendungszweck der Produkte und Risikoklasse müssen die Hersteller eine benannte Stelle ermitteln, die in der Lage sein könnte, sie zu prüfen und ihre Produkte zertifizieren zu lassen. Für IVD der höchsten Risikoklasse (Klasse D) ist zusätzlich zur Einschaltung einer benannten Stelle (NB) oder einer zuständigen Behörde (CA) ein EU-Referenzlabor oder ein Expertengremium erforderlich, um die Leistungsangaben zu überprüfen. Derzeit gibt es nur sechs benannte Stellen, die im Rahmen der EU-IVDR benannt wurden. Warten Sie nicht mit dem Beginn Ihres Antragsverfahrens, um unerwartete Verzögerungen aufgrund der Nichtverfügbarkeit einer benannten Stelle zu vermeiden.

Abbildung 3: Liste der benannten Stellen gemäß IVDR[6]
4. Einrichtung eines Qualitätsmanagementsystems (QMS)
Von IVD-Herstellern wird erwartet, dass sie in ihren Betrieben ein robustes und zuverlässiges Qualitätsmanagementsystem (QMS) einrichten. Dies ist eine allgemeine Verpflichtung des Herstellers gemäß Artikel 10 der IVDR. Das Qualitätsmanagementsystem ist eine wesentliche Anforderung unter vielen anderen, ohne die ein Hersteller nicht zugelassen werden kann.
QMS is to ensure that manufacturing, change control, customer complaints, resource management, supplier &sub-contractors’ controls and validation, performance evaluation, quality test, UDI Labelling, Post market surveillance etc. are according to approved QMS and Post Market Surveillance (PMS) plans.
Das PRRC muss sicherstellen, dass der Hersteller die Anforderungen von Artikel 10 für die "Selbstzertifizierung" (Ausstellung einer Konformitätserklärung gemäß Anhang IV) von IVD der Klasse A erfüllt hat, wenn eine benannte Stelle (NB) in diesem Prozess nicht erforderlich ist.
5. Auf Unterbrechungen in der Lieferkette vorbereitet sein
Throughout the world, manufacturer depends largely on their supply chain and raw material to produce and deliver IVDs that are safe, accurate, and effective forthe intended use. Hence regulatory and quality concerns are also evolving to a higher level when it comes to the suppliers and sub-contractors’ controls. Manufacturer are therefore expected to proactively communicate the supply chain about their obligations and responsibilities of the suppliers and subcontractors. Legal manufacturer shall demonstrate adequate supplier control and monitoring, assure the supply chain is in compliance to the regulatory aspects of IVDR, reconsider the need for data integrity and quality of supplier data, implement robust supplier risk management and performance monitoring and periodically audit the supplier based on the associated risk to the finished products. Die Aufsichtsbehörden und die benannten Stellen fordern die gesetzlichen Hersteller auf, das Niveau der Lieferantenkontrollen klar zu dokumentieren und nachzuweisen, dass sie das Risiko des vom Lieferanten gelieferten Produkts oder der erbrachten Dienstleistung mindern können.
6. Sicherstellung der Audit- und Inspektionsbereitschaft
Gemäß Artikel 88 der IVDR, Marktüberwachungsaktivitäten, führen die zuständigen Behörden sowohl angekündigte (unangekündigte) Inspektionen in den Räumlichkeiten von Wirtschaftsakteuren als auch von Lieferanten und/oder Unterauftragnehmern und, falls erforderlich, in den Einrichtungen von gewerblichen Anwendern durch. Der Hersteller muss Informationen zur Identifizierung aller Standorte, einschließlich der Zulieferer und Unterauftragnehmer, an denen Fertigungstätigkeiten durchgeführt werden, in die technische Dokumentation der Konstruktions- und Herstellungsinformationen aufnehmen. Benannte Stellen (BS), die ein QMS-Audit durchführen, müssen die Verbindungen zwischen den verschiedenen Fertigungsstandorten und deren Zulieferern und/oder Unterauftragnehmern sowie die Verteilung der Verantwortlichkeiten auf diese Standorte ermitteln. Diese Informationen werden berücksichtigt, wenn die BS speziell einen dieser Zulieferer oder Unterauftragnehmer oder beide auditieren will. Die Räumlichkeiten der Zulieferer des Herstellers werden von der BS im Wesentlichen auditiert, wenn davon ausgegangen wird, dass sie die Konformität der fertigen Produkte wesentlich beeinflussen (insbesondere wenn der Hersteller keine ausreichende Kontrolle über seine Zulieferer nachweisen kann).
7. Plan zum Umgang mit unangekündigten Audits
Im Rahmen der Überwachung nach der Zertifizierung führt die BS unangekündigte Vor-Ort-Audits bei den Herstellern und ihren Unterauftragnehmern oder Lieferanten durch, die Produkttests durchführen, und überwacht die Einhaltung aller Bedingungen, die für die Hersteller verbindlich sind und mit den Zertifizierungsentscheidungen zusammenhängen, wie z. B. die Aktualisierung der klinischen Daten in festgelegten Abständen.Außerdem ist tDie benannte Stelle führt mindestens einmal alle fünf Jahre nach dem Zufallsprinzip unangekündigte Audits am Standort des Herstellers und gegebenenfalls am Standort der Zulieferer und/oder Unterauftragnehmer des Herstellers durch, die mit der regelmäßigen Überwachungsbewertung kombiniert werden können.
8. Verstärkung der Überwachung nach dem Inverkehrbringen
Den Herstellern wird dringend empfohlen, ihre Anforderungen an die Überwachung nach dem Inverkehrbringen zu verschärfen und Mechanismen zur Koordinierung der Vigilanz und Marktüberwachung zwischen den EU-Mitgliedstaaten zu entwickeln. Im Rahmen der Überwachungsbewertung für Produkte der Klassen C und D (Anhang IX) führt die benannte Stelle regelmäßig, mindestens einmal alle 12 Monate, geeignete Audits und Bewertungen durch. Dazu gehören auch Audits in den Räumlichkeiten des Herstellers und gegebenenfalls der Lieferanten und/oder Unterauftragnehmer. Der Hersteller muss im Wesentlichen ein Verfahren zur Aufzeichnung und Meldung von Vorkommnissen und Sicherheitskorrekturmaßnahmen im Feld (FSCA) entwickeln.
9. Eindeutige Gerätekennzeichnung (UDI) & EUDAMED
Die Hersteller müssen ein UDI-System einrichten, um die Rückverfolgbarkeit von Produkten zu erleichtern. Um die Rückverfolgbarkeit auf dem EU-Markt zu verbessern, müssen die Etiketten mit einer "Gerätekennung" und einer "Produktionskennung" versehen sein, wobei man sich auf eine Liste akkreditierter Vergabestellen wie GS1, HIBCC, ICCBBA und IFA GmbH beziehen kann, die ein System für die Vergabe von UDIs betreiben. Derzeit gelten die genannten IEs vom 27.th Juni 2019, aber es ist ratsam, ihre Gültigkeit zu bestätigen, bevor eine endgültige Entscheidung über ihre Umsetzung getroffen wird.
Die Europäische Datenbank für Medizinprodukte (EUDAMED) wird einen Überblick über alle in der Europäischen Union erhältlichen Medizinprodukte bieten. Sie besteht aus sechs Modulen zu folgenden Themen:
- Registrierung als Schauspieler,
- Eindeutige Produktkennzeichnung (UDI) und Produktregistrierung,
- Benannte Stellen und Zertifikate,
- Klinische Untersuchungen und Leistungsstudien,
- Vigilanz und Überwachung nach dem Inverkehrbringen, und
- Marktüberwachung.
Um eine verbesserte Transparenz durch ein umfassendes EUDAMED zu gewährleisten, werden Teile der Informationen der Wirtschaftsakteure öffentlich zugänglich sein. Während vertrauliche Informationen nur für Wirtschaftsbeteiligte, Sponsoren, benannte und zuständige Behörden der EU-Mitgliedstaaten zugänglich sind.
10. Anforderungen an "hauseigene Geräte"
Health institution developing ‘in-house devices’ (or ‘laboratory-developed tests’) which are meant to be used by the same health institution shall not be marketed or sold to other legal entity. Such devices may be used for the diagnosis and treatment, especially for rare diseases. The institution is expected to comply with only the requirement of Annex I of IVDR (general safety and performance requirements), and exempted from rest of the regulation until 26 May 2024; provided the health institution meets a number of conditions set out in Article 5(5) of the Regulation and has an appropriate quality management system, which complies to the international standard setting out the quality and competence requirements for medical laboratories (EN ISO 15189) or other national provisions, and is able to justify that target patient group’s specific needs cannot appropriately be met by an equivalent device available on the market.
Referenzen
[1] Offizielle Pressemitteilung der EG vom 20.th Dezember 2021, Schrittweise Einführung der Verordnung über In-vitro-Diagnostika. Abrufbar unter https://ec.europa.eu/commission/presscorner/detail/en/IP_21_6965 [2]VERORDNUNG (EU) 2017/746 DES EUROPÄISCHEN PARLAMENTS UND DES RATES vom 5. April 2017 über In-vitro-Diagnostika und zur Aufhebung der Richtlinie 98/79/EG und des Beschlusses 2010/227/EU der Kommission; ANHANG VIII EINSTUFUNGSVORSCHRIFTEN, 1. DURCHFÜHRUNGSVORSCHRIFTEN Punkt 1.4 Seite 304 [3]Verordnung (EU) 2017/746 des Europäischen Parlaments und des Rates vom 5. April 2017 über In-vitro-Diagnostika und zur Aufhebung der Richtlinie 98/79/EG und des Beschlusses 2010/227/EU der KommissionANHANG IX Konformitätsbewertung auf der Grundlage eines Qualitätsmanagementsystems und der Bewertung der technischen Dokumentation, Seite 306, ANHANG X Konformitätsbewertung auf der Grundlage einer Baumusterprüfung, Seite 314, ANHANG XI Konformitätsbewertung auf der Grundlage einer Qualitätssicherung Produktion, Seite 317
[4] Pressemitteilung dtd. 14. Oktober 2021, Brüssel; Öffentliche Gesundheit: Kommission schlägt schrittweise Einführung der neuen Verordnung über In-vitro-Diagnostika vor [5]Die Auswirkungen der neuen europäischen IVD-Klassifizierungsregeln auf die Beteiligung der benannten Stelle: eine Studie über die in den Niederlanden registrierten IVDs; van Drongelen A, de Bruijn A, Pennings J, van der Maaden T 32 S. in Englisch 2018, RIVM Briefbericht 2018-0082 [6] Die obige Liste basiert auf Daten, die am 5. März 2021 abgerufen wurden. 5. März 2021. Aktuelle Informationen über die Liste finden Sie auf der offiziellen Website der Europäischen Kommission unter https://ec.europa.eu/growth/tools-databases/nando/index.cfm?fuseaction=directive.notifiedbody&dir_id=35Kolabtree helps businesses worldwide hire freelance scientists and industry experts on demand. Our freelancers have helped companies publish research papers, develop products, analyze data, and more. It only takes a minute to tell us what you need done and get quotes from experts for free.

Unlock Corporate Benefits
• Secure Payment Assistance
• Onboarding Support
• Dedicated Account Manager
Sign up with your professional email to avail special advances offered against purchase orders, seamless multi-channel payments, and extended support for agreements.


