



Computer-Biologe und Kolabtree Freiberufler Shaurya Jauhari writes about current challenges involved in conducting pathway analysis in bioinformatics and possible solutions to the problem.
In the contrasting era of “Imprecise Medicine” (propelling us to tune towards Precision Medicine) and inundation of biomedical data brought forth by advancements in instrumentation technologies, a lacuna persists that is largely premised over mapping data to information. The clinical experiments engender Biomarker (technically list of genes or genomic regions more contemporarily) that have to be expounded for their biological implications. The current suite of tools that facilitate such an endeavor is less purposeful as it neglects the ipso factoDas Problem der räumlichen Interaktionen des Genoms angesichts seines Unterbringungsprofils im Kern jeder eukaryontischen Zelle. In diesem Kommentar geht es darum, die Natur des Problems hervorzuheben, die Organisation des Genoms zu beleuchten, kurz über die derzeitigen Kartierungsinstrumente nachzudenken und Vermutungen über mögliche Lösungen anzustellen.
Der Teufel steckt im Detail
Eine ganze Reihe von Bemühungen um eine bessere Auflösung der genomischen Daten streift ein entscheidendes Informationsschema. Wir sind bestrebt, die Wahrscheinlichkeit eines $1000-Genoms voranzutreiben, auch wenn wir uns weniger um die $100.000-Analyse kümmern. Es gibt ein großes Kompendium von Repertoires, die die Anmerkungen zu den experimentellen Ergebnissen und den Fällen unter einer typischen biologischen Studie enthalten. Es könnte Definitionen geben, die die biologischen Implikationen eines Gens oder des Weges, an dem diese Gene beteiligt sind, mit einer Krankheit in Verbindung bringen. Auch diese Art von dynamischen Informationsspeichern wurde (früher) manuell kuratiert, und das Wissensmanagement wurde von automatisierten Pipelines übernommen, die Computer und IKT im Allgemeinen einsetzen. Diese Datenbanken werden mit einer konsensorientierten wissenschaftlichen Weisheit aktualisiert und haben seit ihrer Gründung eine Handvoll Revisionen erfahren. Die Verbindung zwischen den experimentellen Ergebnissen und ihren biologischen Implikationen ist sehr schwach ausgeprägt, vor allem weil die zugrunde liegende "wahre" Biologie außer Acht gelassen wird.
Unser Genom, das im Durchschnitt etwa 2 Meter lang ist, ist im Kern jeder einzelnen Zelle unseres Körpers untergebracht. Aufgrund der winzigen Größe einer Zelle und vor allem ihres Kerns ist das Genom etwas eng und matschig gepackt. Dies ermöglicht es, dass Regionen im Genom, die aus einer linearen Perspektive eher weit voneinander entfernt sind, nahe beieinander liegen und miteinander interagieren. Dieses Sprichwort wird von den derzeitigen Anreicherungswerkzeugen (Kartierungswerkzeugen) grob missachtet, so dass die daraus resultierenden Ergebnisse unverhältnismäßig sind.
Die Regionen im Genom sind Teil von größeren "Aktionsgruppen" oder Pfade that are technically series of chemical reactions accounting for a phenotype; healthy or diseased. When a diseased state is examined, the investigators are on the lookout for the biomarkers that have potentially gone awry and have apparently transformed the organismal body from getönt zu zuckte. Stellen Sie sich vor, Sie jagen eine hart erkämpfte Krankheit mit falsch eingestellten Informationen.
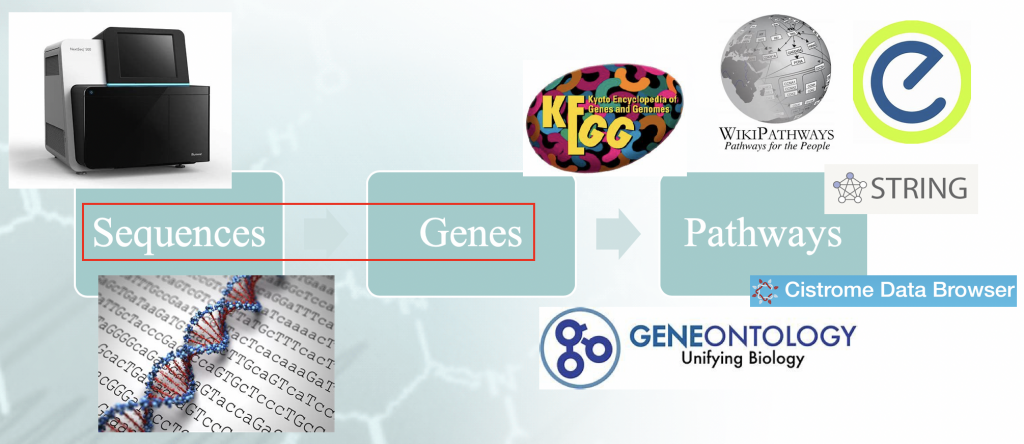 Problembeschreibung - Ablauf einer typischen Anreicherungsanalyse. Es gibt eine bestimmte
Problembeschreibung - Ablauf einer typischen Anreicherungsanalyse. Es gibt eine bestimmte
"Idiomatismus", der mit der Zuordnung der Genomsequenzen zu den Genen verbunden ist und der die
nachgelagerte Ergebnisse.
Genom-Organisation
Wie bereits angedeutet, ist das Genom lang genug, um linear im Zellkern jeder Zelle unseres Körpers oder jedes anderen lebenden Organismus gespeichert zu sein. Vielmehr ist dieses 2 Meter lange Gebilde in eine scheinbar willkürliche Struktur gequetscht und gezwängt, mit Schleifen, Windungen und Wirbeln, wie man sich vorstellen kann. Diese endlos Eine Kette von Nukleotiden oder Basenpaaren - Adenin (A), Cytosin (C), Thymin (T) und Guanin (G) - strukturiert unterschiedliche Topologien innerhalb des Zellkerns und passt sich dabei der Exiguität an. Sie bilden Chromatinschleifen, Kompartimente/Unterkompartimente, Domänen/Unterdomänen, die je nach Zelltyp einen bestimmten Zweck erfüllen. (Beachten Sie, dass verschiedene Zelltypen unterschiedlich funktionieren; eine Nervenzelle hat andere Aufgaben zu erfüllen als eine Muskelzelle; jede Zelle hat eine exklusive Rolle zu spielen).
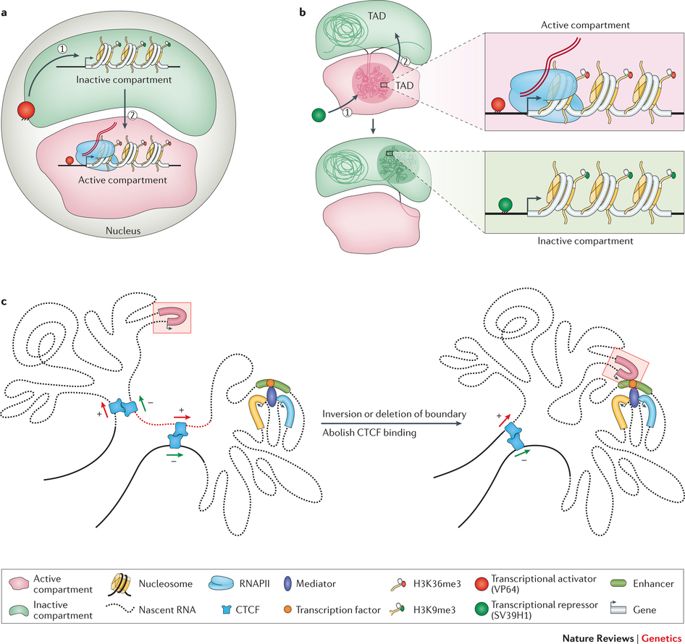
Das Genom windet sich und breitet sich in objektiven Räumen aus. (Kredit: https://doi.org/10.1038/nrg.
2016.112)
Pathway-Datenbanken
Es gibt verschiedene Ontologien und Datenbanken, von denen die Kyoto Encyclopedia of Genes and Genomes (KEGG) (https://www.genome.jp/kegg/) und der Gene Ontology (http://geneontology.org/Die wenigen Tools, die ich im nächsten Abschnitt vorstellen werde (und die oft meinungsbildend sind), erzeugen Anreicherungsbegriffe, die "selektiv" aus den genannten Datenbanken stammen. Anhand ihrer statistischen Signifikanzwerte wird abgeleitet, ob sie wirklich einen aufgeführten Phänotyp repräsentieren oder ob es sich nur um eine zufällige Entwicklung handelt. (P.S. Es gibt eine Abhandlung über p-Werte, die vermutlich Laien helfen wird, die Idee der p-Werte zu verstehen. statistische Signifikanz. Bitte folgen Sie dem Link https://sway.office.com/WkyHrPnVB8Ec3zPD?play nach eigenem Ermessen).
Anreicherungstools
Die Anreicherungsanalyse ist ein Berechnungsprotokoll, das novo genomischen Regionen mit ihren Definitionen in den erwähnten Datenbanken zu vergleichen. Was die Instrumente (die als Kanal dienen) betrifft, so werden sie klassischerweise in mehrere Kategorien unterteilt, nämlich in die Überrepräsentationsanalyse (ORA), die Funktionsklassenbewertung (FCS), die auf der Pfad-Topologie (PT) basierenden Methoden und die Netzwerk-Interaktionsmethoden (NI).
Analyse der Überrepräsentation
Bei der Überrepräsentationsanalyse wird anhand des Dogmas der hypergeometrischen Verteilung die Menge der unterschiedlich exprimierten Gene auf diejenigen untersucht, die Teil eines biologischen Weges sein könnten. Ein hypergeometrischer Test berücksichtigt im Wesentlichen vier Attribute, um eine Entscheidung zu treffen, nämlich.
- Gesamtzahl der Gene in dem betreffenden Assay,
- Die differenziell exprimierten Gene,
- Anteil der Gene im Zielpfad an der Gesamtzahl der Gene, und
- Unterschiedlich exprimierte Gene, die im Zielpfad vorkommen.
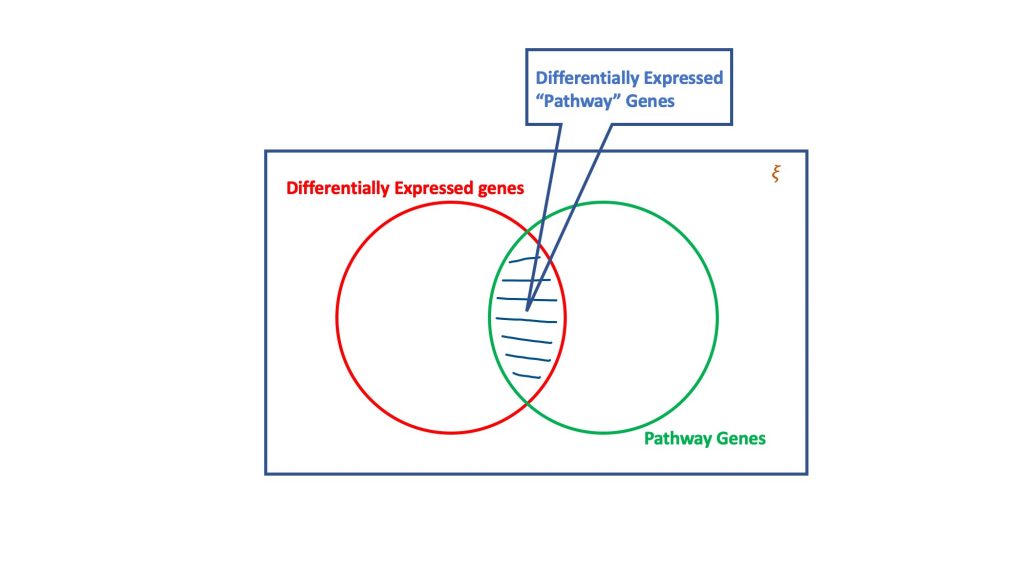
Das Wesen des hypergeometrischen Tests
Despite being a simple and straightforward methodology, ORA presents its own limitations.
- Demokratie im Spiel; alle Gene werden gleich berücksichtigtWarum ist das ein Problem? Nehmen wir an, dass die Gene auf der Grundlage folgender Kriterien gefiltert werden Fold-change. Wir sichten Gene, die Expressionsunterschiede von mehr als oder gleich 2 Mal (Falten) aufweisen, sowohl in negativer als auch in positiver Richtung. Obwohl das Minimum das 2-fache war, würde dieser Arbeitsablauf auch Gene mit Ausdrucksänderungen erfassen, die 3-fach, 4-fach und mehr sind. Sicherlich ist ein Gen mit einer 4-fachen Expressionsabweichung umsichtiger als ein Gen mit einer 2-fachen Veränderung. Diese Erscheinung wird von ORA außer Acht gelassen.
- Berücksichtigt nur die wichtigsten GeneNehmen wir noch einmal ein Gen mit einer Fold-change von 1,9999 oder einem p-value <0,0051113; üblicherweise wird ein p-value <0,05 als statistisch signifikant angesehen. Bei der ORA-Methode wird dieses Gen im Endergebnis übergangen. Es ist klar, dass es einen Informationsverlust und einen Mangel an Flexibilität gibt. (P.S. Breitling et al. haben sich mit diesem Dilemma befasst, indem sie eine Überarbeitung zur Vermeidung von Schwellenwerten vorschlugen. Bei der Überarbeitung wird ein iterativer Ansatz verwendet, bei dem ein Gen nach dem anderen hinzugefügt wird, um eine Reihe von Genen zusammenzustellen, für die ein Signalweg optimal signifikant ist).
- Kein Gen funktioniert isoliertDies ergibt sich aus den oben erwähnten Einschränkungen, dass die Behandlung von Genen als unabhängige Einheit den Kern des polygenen Beitrags zu einem Phänotyp aus den Augen verliert. Ein Ziel der Genexpressionsanalyse könnte darin bestehen, Genkohorten zu ermitteln, deren Expressionsmuster kongruent sind. Diese Symphonie hebt funktionell verwandte Gene oder Gene, die auf einen gemeinsamen biologischen Zustand hinarbeiten, hervor.
- Gegenseitig unabhängige PfadeDie ORA geht auch davon aus, dass die Wege nicht hintereinander (oder nacheinander) ablaufen. Dies ist ein prinzipieller Fehler, da eine Reihe von chemischen Reaktionen durchaus vor- oder nacheinander ablaufen kann.
Bewertung der Funktionsklasse
Contrary to ORA, FCS methods subsume all the contextual genes as well as their association Statistik (fold-change, p-value) and compute a läuft Anreicherungswert für Gengruppierungen (auf der Grundlage von funktionellem Wissen wie Gene Ontology oder KEGG-Pfade), z. B. GSEA vom Broad Institute (http://software.broadinstitute.org/gsea/index.jsp). Bei einem typischen FCS-Lauf wird die Veränderung der Expression aller Gene in der Liste der unterschiedlich exprimierten Gene in einem Experiment analysiert (nicht nach statistischer Signifikanz oder ähnlichem geordnet). Das Hauptergebnis der Analyse der Anreicherung von Gensätzen ist ein Anreicherungswert (ES), der das Ausmaß widerspiegelt, in dem ein Gensatz am Anfang oder am Ende einer Rangliste von Genen überrepräsentiert ist; warum am Anfang und am Ende? weil dies die Gene sind, die in Bezug auf die Expressionsänderung am weitesten von der Norm entfernt sind. Ein positiver ES-Wert für einen Gensatz (oder einen Zielpfad, GS) auf die Gene in der Liste hinweisen (GL), die oben liegen (am stärksten hochreguliert; 1,2,3 ...), während ein negativer ES-Wert bedeutet, dass die Komponentengene unten liegen (am stärksten herunterreguliert; n-3, n-2, n-1, n, wobei n die Gesamtzahl der Gene ist). P.S. Der ES-Wert wird zum normalisierten ES-Wert (NES), wenn das Problem der Mehrfachtests korrigiert wird (Falschentdeckungsrate, z. B. Bonferroni-Methode).
Zusammenfassend lässt sich sagen, dass die FCS-Methoden den ORA-Methoden um einiges überlegen sind,
- Verzicht auf die Festlegung eines willkürlichen Schwellenwerts für die Einstufung von Genen als signifikant oder nicht signifikant.
- die Auswertung von Informationen über die Genexpression, um systematische Veränderungen in den Stoffwechselwegen zu verfolgen; dadurch wird die Verantwortlichkeit für die gegenseitige Abhängigkeit der Gene deutlich.
Allerdings haben auch die FCS-Methoden gewisse Schwächen.
- Da die Pfade unabhängig voneinander analysiert werden, werden Gene, die mehrere Pfade regulieren, möglicherweise nicht gezählt.
- Viele FCS-Methoden ordnen die Gene in einer Liste auf der Grundlage der Veränderungen in der Genexpression. Ein Szenario, in dem der Unterschied in den Rängen eine ungleiche (und möglicherweise exponentielle) Varianz in der Expression widerspiegelt, könnte vielleicht ein unfaires Maß sein.
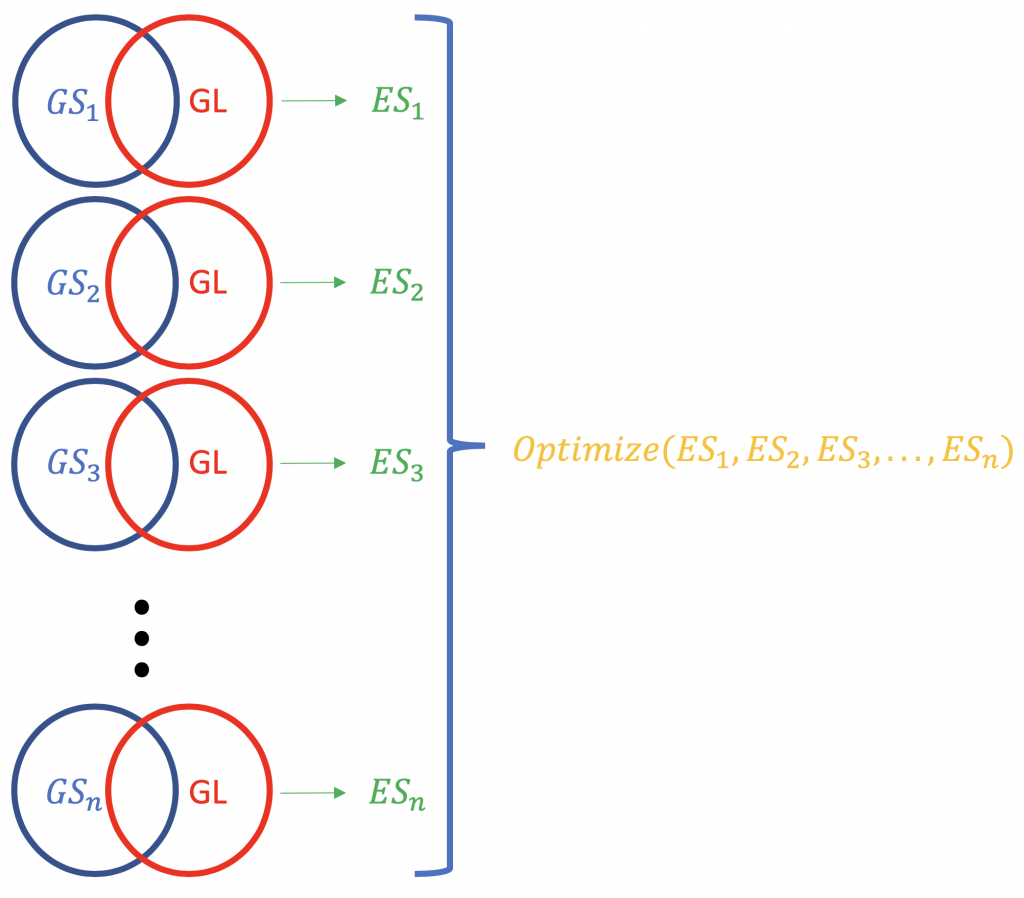
Die Normalisierung der ES-Scores für eine Genliste.
Auf Pfad-Topologie basierende Ansätze
Ein entscheidender Nachteil der ORA- und FCS-Methoden besteht darin, dass sie die Struktur der Signalwege ignorieren. Die Reihenfolge der Gene, die in einem Signalweg reguliert werden, ist für die Rückverfolgung der kausalen Auswirkungen von wesentlicher Bedeutung. Es ist verständlich, dass es genau zwei Pfade mit denselben Genkomponenten geben kann, aber die Hierarchie der Aktivierung könnte völlig unterschiedlich sein. Wäre es nach den ORA/FCS-Methoden gegangen, hätten sie zu ähnlichen Anreicherungsbedingungen geführt. Das ist ein Problem. Pathway Topology(PT)-Methoden gehen von einer ausschließlichen Funktion in Abhängigkeit von den spezifischen Interaktionen aus, was auch der allgemeinen Logik entspricht. Beispiele für Werkzeuge sind SPIA (https://www.bioconductor.org/packages/release/bioc/manuals/SPIA/man/SPIA.pdf), GGEA, und PARADIGM. Im Allgemeinen haben die Tools dieser Kategorie einen lokalen und einen globalen Score. Der lokale Score auf Genebene kalibriert die Fold Changes in der Expression des Gens und der vorgelagerten Gene, während der globale Score das Maß für die Verwandtschaft mit dem Gensatz auf Wegebene misst. Dies führt jedoch auch dazu, dass PT-Methoden die Daten für einen bestimmten Zustand/Zelltyp überanpassen.
Auf Netzwerkinteraktion basierende Analyse
Dies ist eine eher unterschätzte Kategorie, die trotz ihrer veralteten Formulierung noch kaum umgesetzt wird. Methoden wie EnrichNet , NetPEA (http://www.dx.doi.org/10.1109/BIBM.2013.6732493) have been proposed close to a decade ago, but they haven’t gained much traction because of limited tools available. This facade constraints the dexterity of the theme as no improvements are documented. That turns out to be an open-ended Forschung problem.
Der offensichtliche Allheilmittel
Wahrscheinlich haben Sie jetzt eine Vorstellung von der grundlegenden Anreicherungs-/Pfadanalyse und der Art der Tools, die dabei helfen. Wie ich bereits erwähnt habe, stützen sich jedoch alle verfügbaren Tools (die in die markierten Kategorien fallen) auf einen schiefen Parameter eines linearen Fensters über der abgefragten Region. Wenn die dazwischen liegenden Segmente des Genoms in diesen Rahmen fallen, werden sie als angereichert, sonst nicht. Es geht darum, ein Werkzeug zu haben, das vielleicht eine Zentrum und Durchmesser eines hypothetischen Kreises, um sozusagen die 3D-Organisation auf der Grundlage der Genominteraktionen für die Region hervorzuheben.

GREAT bietet mehrere erweiterte Optionen, um die lineare Dimension um die Transkriptionsstartstelle eines Gens zu spezifizieren.
 Enrichr bietet eine unkomplizierte Auswahl des Genomtyps und der Anzahl der Gene in der vermuteten linearen Region.
Enrichr bietet eine unkomplizierte Auswahl des Genomtyps und der Anzahl der Gene in der vermuteten linearen Region.
Wie aus Abbildung 4 hervorgeht, gibt es kein Verständnis für die "wahre" räumliche Organisation des Genoms. Dies ist ein Problem und eine tiefe Verwerfung, die im derzeitigen Bereich der Anreicherungsanalyse fortbesteht. Ungeachtet dessen gibt es auch eine starke Relevanz von TranskriptionsfabrikenDie Transkriptionsfabriken, die als Orte im Kernraum identifiziert wurden, die entfernte regulatorische Elemente zur "Party im Haus" locken. Scherzhaft bemerke ich oft, dass, ähnlich wie jemand, der wütend auf jemanden oder etwas ist, normalerweise sagen würde, Transkriptionsfabriken (personifiziert) würden das Genom möglicherweise zurechtweisen - "Die Abschrift wird nur über meine Leiche erfolgen, sonst nicht!". Das angrenzende Thema der Transkriptionsfabriken ist Gegenstand einer weiteren, zukünftigen Diskussion. Es verdeutlicht jedoch das Dogma der cis-regulatorischen Interaktionen, das in der heutigen Praxis nicht mehr stimmt.
Abschließend möchte ich anmerken, dass die Pfadanalyse ein entscheidender und oft vernachlässigter Teil der Forschung ist. Bioinformatik Pipeline. Es gibt immer einen Spielraum für die Skalierung der bestehenden Methoden in Verbindung mit den Genomdaten, die sich in diesem Moment weiterentwickeln. Wenn uns ein größeres Datenvolumen zur Verfügung steht, wird dies nicht nur ein infrastrukturelles Problem sein, sondern auch ein algorithmisches Problem.
Notwendigkeit einen Bioinformatik-Berater einstellen? Arbeiten Sie mit freiberuflichen Wissenschaftlern auf Kolabtree. Es ist kostenlos, Ihr Projekt zu veröffentlichen und Angebote von Experten zu erhalten.
Verwandte Experten:
Bioinformatik Freiberufler | Pflanzengenetik | Entwicklungsbiologie | Gentherapie | Stammzellen |
Analyse von DNA-Sequenzierungsdaten |Tiergenetik | Wechselwirkungen mit Medikamenten | Genetik und Genomik
Kolabtree helps businesses worldwide hire freelance scientists and industry experts on demand. Our freelancers have helped companies publish research papers, develop products, analyze data, and more. It only takes a minute to tell us what you need done and get quotes from experts for free.

Unlock Corporate Benefits
• Secure Payment Assistance
• Onboarding Support
• Dedicated Account Manager
Sign up with your professional email to avail special advances offered against purchase orders, seamless multi-channel payments, and extended support for agreements.


