



Computational biologist and Kolabtree freelancer Shaurya Jauhari writes about current challenges involved in conducting pathway analysis in bioinformatics and possible solutions to the problem.
In the contrasting era of “Imprecise Medicine” (propelling us to tune towards Precision Medicine) and inundation of biomedical data brought forth by advancements in instrumentation technologies, a lacuna persists that is largely premised over mapping data to information. The clinical experiments engender biomarkers (technically list of genes or genomic regions more contemporarily) that have to be expounded for their biological implications. The current suite of tools that facilitate such an endeavor is less purposeful as it neglects the ipso facto, spatial interactions of the genome given its accommodation profile inside the nucleus of every eukaryotic cell. This commentary is about highlighting the nature of the problem, shedding light on genome organization, reflecting briefly on the present mapping tools, and conjecturing the possible solutions.
Devil is in the Detail
A bevy of efforts towards honing resolution of the genomic data is brushing underneath a crucial scheme of information. We’re anxiously impelling the likeliness of a $1000 genome, albeit care less of the $100,000 analysis. There exists a large compendium of repertoires that hold the annotations to the experimental results and the cases under a typical biological study. There could be definitions stating the biological implications of a gene, or the pathway those genes are a part of, being associated with a malady. Again, these kind of dynamic, informational stores have been manually curated (erstwhile) and the knowledge management has been taken over by automated pipelines employing computers and ICT at large. These databases are updated with a consensus-oriented scientific wisdom and have embraced a handful revisions since inception. The conduit mapping the experimental results to their biological implications is starkly subdued, largely because the underlying “true” biology is dismissed.
Our genome, averaging around 2 meters in length, is accommodated inside the nucleus of every single cell that lines our body. Owing to the diminutive frame of a cell and more-so of it’s nucleus, the genome is packed in somewhat of a strained and squishy manner. What this allows for are regions in the genome, that are rather distant from a linear perspective, coming in close vicinity and interacting. This adage grossly rejected by the current suite of enrichment (mapping) tools and therefore the results engendered are disproportionate.
The regions in the genome are part of larger “action-groups” or pathways that are technically series of chemical reactions accounting for a phenotype; healthy or diseased. When a diseased state is examined, the investigators are on the lookout for the biomarkers that have potentially gone awry and have apparently transformed the organismal body from toned to twitched. Imagine chasing a hard-fought disease with misaligned information.
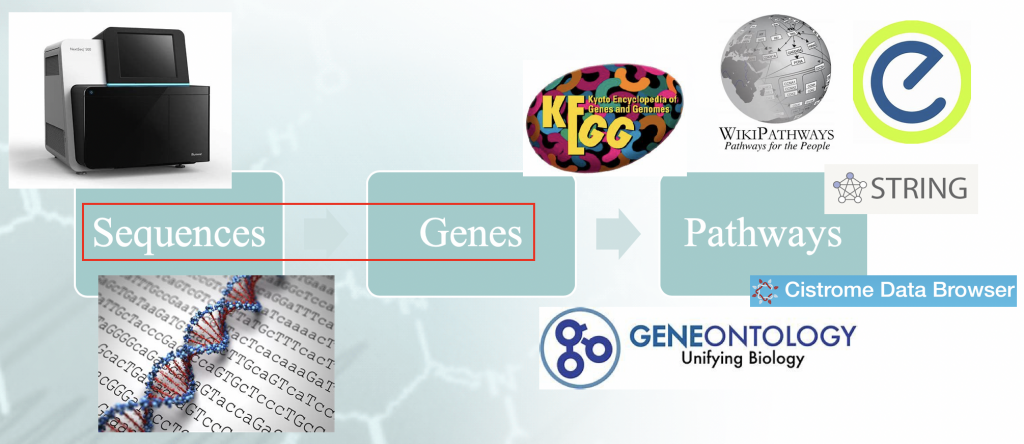 Statement of the Problem — Workflow of typical enrichment analysis. There is a certain
Statement of the Problem — Workflow of typical enrichment analysis. There is a certain
”idiomatism” associated with the mappping of the genome sequences to genes, and that orchestrates the
downstream results.
Genome Organization
As alluded to before, the genome is long enough to be stored linearly inside the nucleus of a cell, for every cell in our body or any other living organism for that matter. Rather this 2 meter entity is squished and crammed into a seemingly haphazard structure, with loops. turns, and swirls as one may imagine. This endless string of nucleotides or base-pairs- Adenine (A), Cytosine (C), Thymine (T), and Guanine(G) structures distinct topologies inside the nucleus while adjusting to the exiguity. They form chromatin loops, compartments/ sub-compartments, domains/ sub-domains that serve a purpose, in accordance to the cell type. (Note that different kind of cells function differently; a nerve cell has other businesses to cater than a muscle cell; each one has a exclusive role to play.)
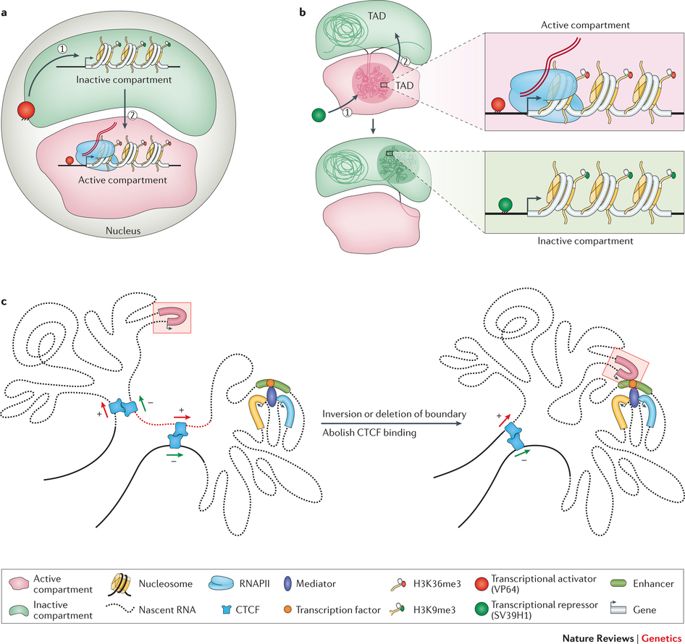
The genome coils and sprawls into objective spaces. (Credit: https://doi.org/10.1038/nrg.
2016.112)
Pathway Databases
Variegated ontologies and databases exist, out of which Kyoto Encyclopedia of Genes and Genomes (KEGG) (https://www.genome.jp/kegg/) and Gene Ontology (http://geneontology.org/) are majorly queried.The few tools that I shall be briefing in the upcoming section (and that are often opinionated) beget enrichment terms that are “selectively” from the said databases. On the basis of their statistical significance values, it is derived whether they truly represent a listed phenotype or are just a matter of random development.(P.S. There is a write-up on p-values that will presumably help laymen understand the idea of statistical significance. Please follow the link https://sway.office.com/WkyHrPnVB8Ec3zPD?play at your convenience).
Enrichment Tools
The enrichment analysis is a computational protocol of novo genomic regions to their recorded definitions in the databases that have been alluded to. Speaking of the tools, (that act as a conduit), they are classically structured under several heads, viz. over-representation analysis (ORA), functional class scoring (FCS), pathway topology (PT) based methods, and network interaction (NI) methods.
Over Representation Analysis
The over-representation analysis, via the dogma of hypergeometric distribution, evaluates the set of differentially expressed genes for the ones that could be part of a biological pathway. Basically, a hypergeomtric test considers four attributes to reach a decision, viz.
- Total number of genes in the considered assay,
- The differentially expressed genes,
- Genes in the target pathway out of the total number of genes, and
- Differentially expressed genes occurring in the target pathway.
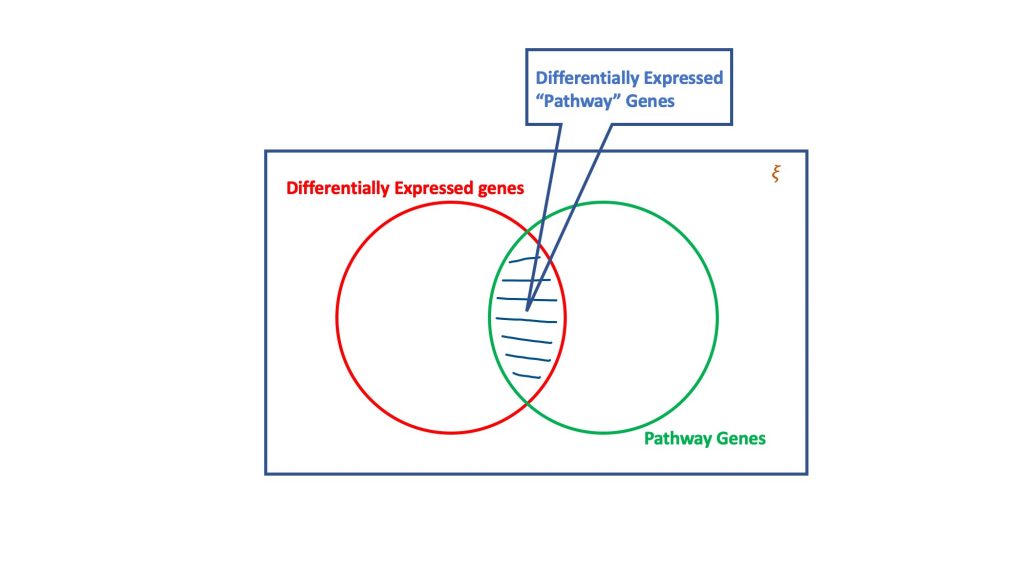
The essence of hypergeometric test
Despite being a simple and straightforward methodology, ORA presents its own limitations.
- Democracy in play; All genes are considered equally; Why is that a problem? Let’s assume that the genes are filtered on the basis of fold-change. We sift genes that embraced expression difference greater than or equal to 2 times(folds), in both negative and positive directions. Albeit, the minimum was 2-fold, this workflow would also capture genes with changes in expression were 3-fold, 4-fold, and beyond. Surely, a gene with 4-fold expression disparity is more prudent than a gene with 2-fold change. This manifestation is discounted by ORA.
- Considers only most significant genes; Again, let us consider a gene with fold-change 1.9999 or p-value <0.0051113; commonly, a p-value <0.05 is considered statistically significant. ORA methodology glosses over this gene in the final result. Clearly, there is a information loss and dearth of flexibility. (P.S. Breitling et al. addressed this predicament by proposing an extemporization for avoiding thresholds. The revision employs an iterative approach that appends one gene at a time to compile a set of genes for which a pathway is optimally significant.)
- No gene works in isolation; This follows from the aforementioned limitations that treating gene as an independent entity loses the nub of the polygenic contribution towards a phenotype. One goal of gene expression analysis could be to elucidate gene cohorts whose expression patterns are congruent. This symphony highlights functionally akin genes or genes working towards a common biological state.
- Mutually independent pathways; ORA also assumes that pathways do not work in tandem (or in succession). This is principally flawed as series of chemical reactions could well precede or proceed one another.
Functional Class Scoring
Contrary to ORA, FCS methods subsume all the contextual genes as well as their association statistics (fold-change, p-value) and compute a running enrichment score for gene groupings (based on some functional knowledge like Gene Ontology or KEGG pathways). eg. GSEA from Broad Institute (http://software.broadinstitute.org/gsea/index.jsp). A typical FCS run would analyse the expression change of overall genes in the list (not ranking by statistical significance or something else) of differentially expressed genes in an experiment. The primary result of the gene set enrichment analysis is an enrichment score (ES) that reflects the degree to which a gene set is overrepresented at the top or bottom of a ranked list of genes; why top and bottom? because there are the genes farthest from the normal, in terms of the expression change. A positive ES score for a gene set (or a target pathway, GS) will be indicative of the genes in the list (GL) falling into the top (most upregulated; 1,2,3 …), while a negative ES score shall mean that the component genes lay on the bottom (most downregulated; n-3, n-2, n-1, n, where n is the total number of genes). P.S. The ES becomes normalised ES (NES) when correcting for multiple testing issue (false discovery rate, eg. Bonferroni method).
In summary, FCS methods are notably better than ORA methods by,
- shunning the requirement of an arbitrary threshold for classifying genes as significant or non-significant.
- appreciating information on gene expression to track systematic changes in the pathway; this renders accountability on gene inter-dependence.
Albeit, FCS methods have certain shortcomings too.
- Since pathways are analyzed independently, genes that regulate several pathways might not be counted.
- Many FCS methods rank the genes in a list on the basis of changes in gene expression. A scenario where the difference in ranks reflects unequal(and possibly exponential) variance in expression could perhaps be an unfair measure.
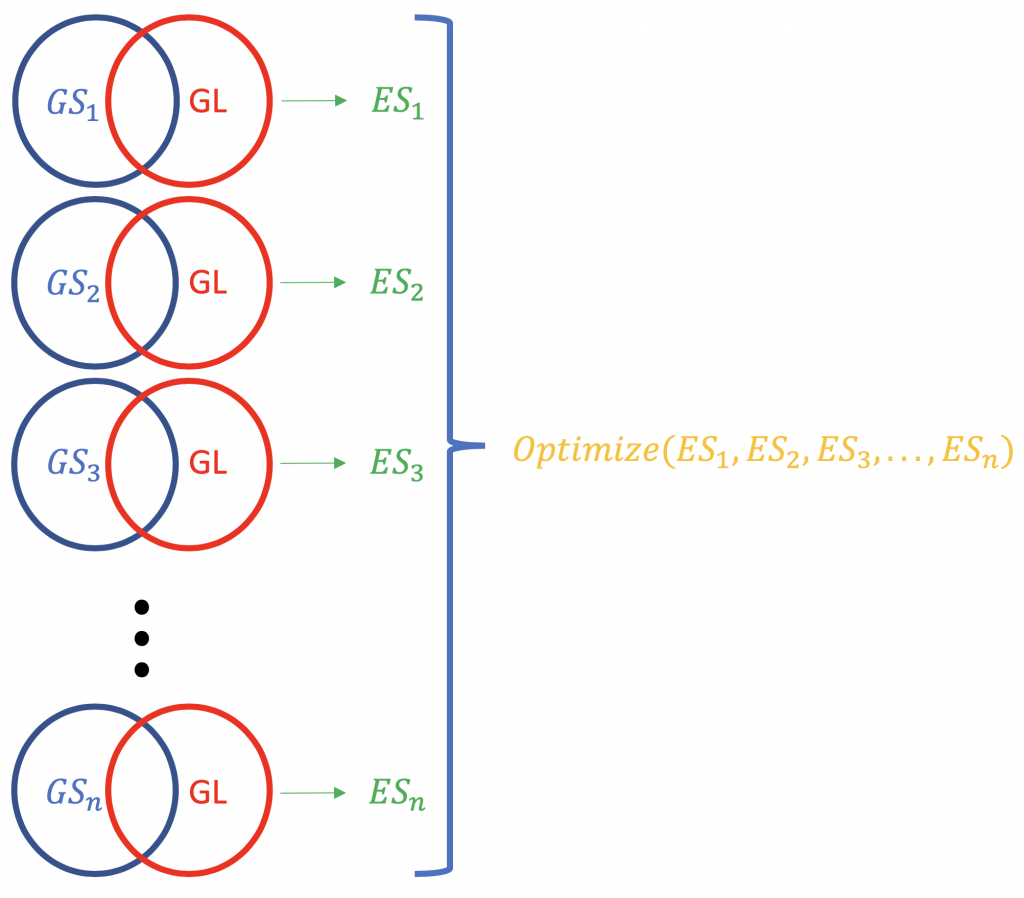
The normalisation of the ES scores pertaining to a gene list.
Pathway Topology based Approaches
A crucial deprivation from the ORA and FCS methods is that they ignore the structure of the pathways. The order of genes that are regulated in a pathway is essential to trace the causal effects. Understandably, there could be exactly two pathways with same gene components but the hierarchy of activation might be entirely different. If it were for the ORA/FCS methods, they would’ve resulted in similar enrichment terms. That is a problem. Pathway Topology(PT) methods assume exclusivity in function depending on the specific interactions, which conforms to the general logic too. Examples for tools are SPIA (https://www.bioconductor.org/packages/release/bioc/manuals/SPIA/man/SPIA.pdf), GGEA, and PARADIGM. Generally, tools of this category shall have a local and a global score. The local score at the gene level shall calibrate fold changes in the expression of the gene and genes upstream, while the global score shall gauge the pathway level measure for the relatedness with the gene set. Nonetheless, this also renders that PT methods overfit the data for a peculiar condition/cell type.
Network Interaction based Analysis
This is rather a understated category that is still scarcely implemented, despite of its dated formulation. Methods like EnrichNet , NetPEA (http://www.dx.doi.org/10.1109/BIBM.2013.6732493) have been proposed close to a decade ago, but they haven’t gained much traction because of limited tools available. This facade constraints the dexterity of the theme as no improvements are documented. That turns out to be an open-ended research problem.
The apparent Panacea
You probably now have a narrative about fundamental enrichment/ pathway analysis and the kind of tools that aid it. However, as I mentioned before, all the tools available (falling across the marked categories) rely on a skewed parameter of a linear window across the queried region. If the intervening segments of the genome fall into this frame, they are listed as enriched, else not. The real deal is to have a tool that, perhaps, inputs a centre and diameter of a hypothetical circle, as it were to highlight the 3D organization based genome interactions for the region.

GREAT offers several advance options to specify linear dimension around the Transcription Start Site of a gene.
 Enrichr presents straightforward selection of genome type and number of genes in the presumed linear region.
Enrichr presents straightforward selection of genome type and number of genes in the presumed linear region.
As inferred from the Figure 4, there is no appreciation for the “true” spatial organization of the genome. This is a problem and a deep fault-line that persists in the current domain of enrichment analysis. Notwithstanding, there is also a stark relevance of transcription factories, identified as sites in the nuclear space that lure distant regulatory elements to “party in-house”. Jokingly, I often remark that akin to someone who is livid with someone or something would usually say, transcription factories (personified) would conceivably reprimand the genome- “The transcription will ensue over my dead body, none elseways!“. The adjacent theme of transcription factories is a subject of some adjacent, future discussion. However, it materializes the dogma of cis-regulatory interactions that is amiss in the contemporary practice.
In closing, I would like to remark that pathway analysis is a crucial, and often neglected fraction of the Bioinformatics pipeline. There is always a scope for scaling the existing methodologies in tandem with the genomic data, which is evolving as we speak. When escalated volume of data becomes available to us, it is not only going to be a infrastructural problem but an algorithmic clampdown too.
Need to hire a bioinformatics consultant? Work with freelance scientists on Kolabtree. It’s free to post your project and get quotes from experts.
Related experts:
Bioinformatics freelancer | Plant genetics | Developmental Biology | Gene therapy | Stem cells |
DNA sequencing data analysis |Animal genetics | Drug interactions | Genetics and Genomics
Kolabtree helps businesses worldwide hire freelance scientists and industry experts on demand. Our freelancers have helped companies publish research papers, develop products, analyze data, and more. It only takes a minute to tell us what you need done and get quotes from experts for free.

Unlock Corporate Benefits
• Secure Payment Assistance
• Onboarding Support
• Dedicated Account Manager
Sign up with your professional email to avail special advances offered against purchase orders, seamless multi-channel payments, and extended support for agreements.


