



A clinical evaluation report (CER) is a technical document required by medical device companies that want to sell or distribute products in Europe. The CER provides a comprehensive overview of the medical device design and composition, intended usage and applications, clinical trials analysis and outcomes, relevant literature reviews, protocols and instructions for use.
EU MDR and CE Marking
Medical devices in Europe need to get a “CE Mark”, which indicates that the product complies with the EU regulations (MDD, MDR or IVDR, whichever is applicable). The recent Medical Device Regulations in Europe has significantly impacted the way medical devices are regulated and approved in the EU. The MDR is the biggest change to medical device legislation in over 20 years. Some major changes in the MDR (as compared with the MDD) include device classification, technical file documentation, traceability and post-market surveillance.
Read more: Top tips to get a CE Mark for your medical device
Clinical Evaluation Report
The MDR places great emphasis on Clinical Evaluation Reports or CERs. The CER forms part of the Technical File, which is the main documentation which demonstrates regulatory compliance and provides all the information about the device. Medical device companies need to take several measures to make sure that their CER is well-prepared and well-maintained. CER documentation typically comprises 4 stages:
- Defining the scope of the device, its intended usage and therapeutic/diagnostic claims
- Identifying and validating clinical data
- Analyzing the data and interpreting it to see if it meets all the requirements
- Identifying risks and uncertainties, which may be answered during post-market surveillance (PMS)
A CER needs to be regularly updated throughout the life cycle of a medical device. The document should contain the clinical evidence that supports proof of conformity to the Essential Requirements (ERs) in MEDDEV 2.7/1 Rev. 4 Annex 1 (Safety and Performance Requirements in the MDR). The document should also describe the physical and technical aspects and composition of the device, along with the instructions for use.
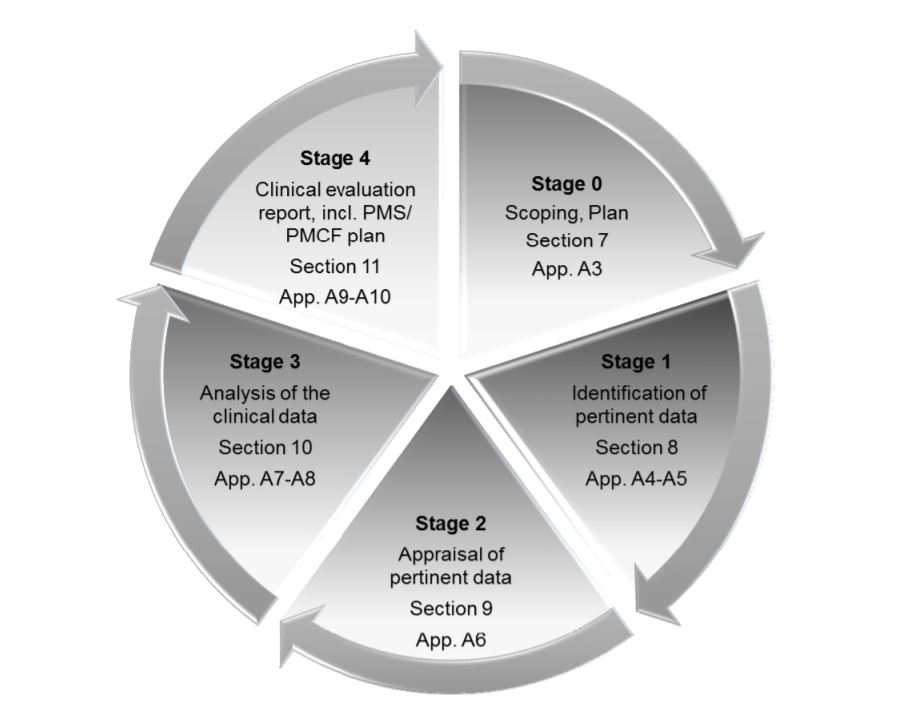
The four stages of CER as outlined in MEDDEV 2.7/ rev. 4.
Equivalence
Medical device manufacturers need to evaluate clinical data from their own medical device or an equivalent device, whose safety and clinical performance is no different. Under the MDR, the medical device manufacturer has to consider three factors to prove a product is equivalent: biological, technical and clinical.
Technical equivalence means that the device must have the same design specifications and composition, and must be used in the same conditions, with the same deployment method and operational principles. Clinical equivalence means that the device must be used to treat the same condition, at the same location in the body and for the same group of population, achieving similar performance.
Biological equivalence means that the medical device in question should not cause any biological risk.
If a medical device is unable to demonstrate equivalence, they have to either perform further clinical investigation or remove the claim from their product.
Literature Review
Literature search results form a core part of the clinical evaluation report.A strong literature review process needs to be established so that all published literature relevant to the device is easily accessible and can support the claim being made. Literature reviews typically need the expertise of a skilled researcher who can identify the correct search term, use databases like PubMED or MEDLine, find relevant literature and appraise it, including using relevant references. The literature review should also address any questions that the clinical data is unable to answer.
How long does it take to write a CER?
Identifying and gathering all the data that goes into the CER and preparing the document according to the guidelines can take several weeks or months. Post-market surveillance (PMS) forms a big part of the process even after CE Mark certification, and PMS has to be a core part of the manufacturers Quality Management System (QMS). Some cases also require Post-Market Clinical Followup (PMCF) which involves proactive collection of clinical data and evaluating it to meet safety and performance requirements.
The CER is not a static document. It is recommended that medical device manufacturers develop a robust process and strategy incorporating good practices to help them retain market market access in the EU, avoid any product recalls, and streamline their time and resources used to maintain the document.
Outsourcing CER writing to a freelance expert can help save precious time and energy. Freelance clinical evaluation report writers are able to develop CERs starting from USD 3,000 on Kolabtree.
Clinical Evaluation Report Writers
The time and expertise required to put together a CER can be a resource strain for several medical device startups or medical device SMBs, who may not have in-house specialists. Working with external experts, including freelance CER writers, clinical data analysts, literature review experts biostatisticians and medical writers can help companies make sure that their CER strategy is robust and their data is all verified and accurate.
How to hire a freelance CER writer
Skills to look for in a CER writer include experience dealing with medical device regulations and approval processes, familiarity with MEDDEV 2.7/ rev. 4, knowledge of therapeutic areas and strong analytical skills. Kolabtree’s global pool of experts include qualified Clinical Evaluation Report writers, who can help you put together a well-researched and thorough CER. It’s free to post your project and get quotes from experts. Get started
Related Experts:
Medical Device Consultants | Freelance Medical Writer | Freelance CER Writer | EU MDR Consultants | FDA Submissions | Literature Search Experts | Regulatory Compliance Experts | Clinical Trials Expert | Regulatory Writers | PMS Consultants | PMCF Consultants | Biostatistics Experts | Product Development Expert
Kolabtree helps businesses worldwide hire freelance scientists and industry experts on demand. Our freelancers have helped companies publish research papers, develop products, analyze data, and more. It only takes a minute to tell us what you need done and get quotes from experts for free.

Unlock Corporate Benefits
• Secure Payment Assistance
• Onboarding Support
• Dedicated Account Manager
Sign up with your professional email to avail special advances offered against purchase orders, seamless multi-channel payments, and extended support for agreements.


