

Sundeep Agarwal, dispositivo médico regulatory consultant on Kolabtree, shares the essential requirements of a FDA 510k premarket notification to ensure success.
Com a corrente $156 bilhão de dólares no mercado, e espera-se $208 bilhões [1] by 2023, the US medical device market is undoubtedly lucrative. Additionally, the aging population, history of chronic diseases, the saúde system and disruption in supply chain encourages global manufacturer to invest and expand their business horizon in the United States. Like any other global manufacturer, if you think you can leave an imprint in the world largest medical device market, you have just hit on the right blog to prepare and understand the regulatory requirement with respect to FDA’s 510(k)for your device which is utmost vital for the application process.
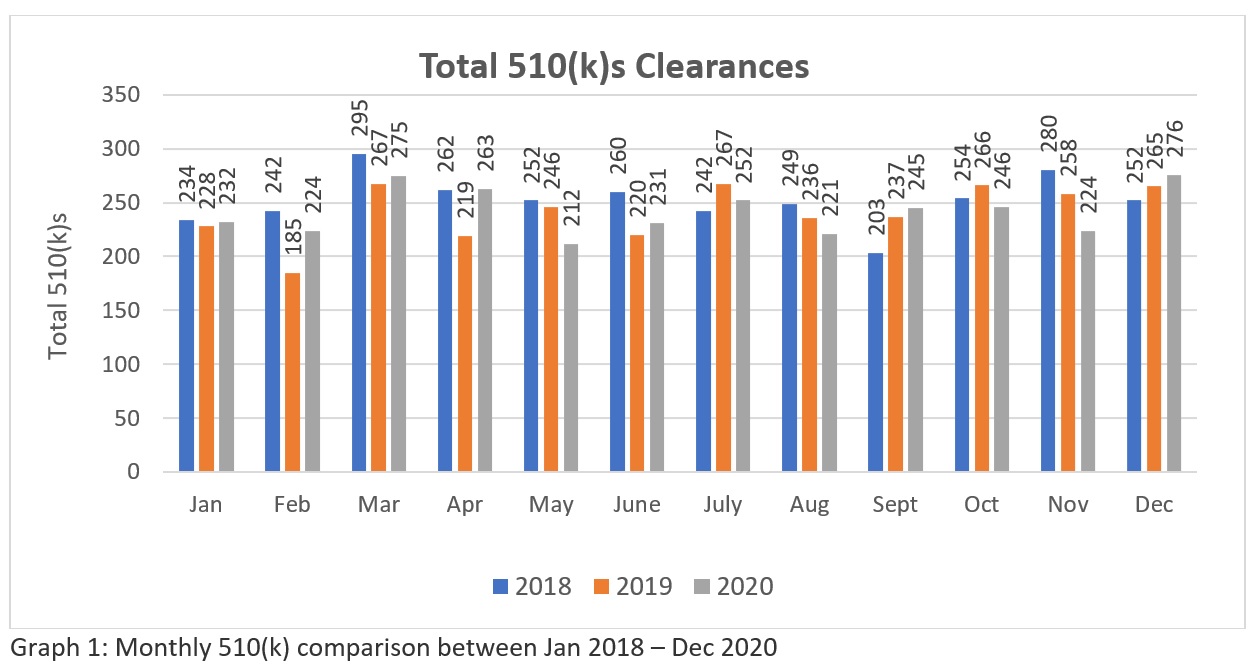
A trend analysis of three years FDA’s 510k clearances data (Source: https://www.fda.gov/medical-devices/device-approvals-denials-and-clearances/510k-clearances) revelam que um total de 3025-510(k)swere clear entre janeiro de 2018 a dezembro de 2018, 2894 - 510(k)s entre janeiro de 2019 a dezembro de 2019 e 2901 - 510(k)s liberados entre janeiro de 2020 e dezembro de 2020. Estes números (Consulte a Tabela 1) sugerem que atualmente, a FDA tem o potencial de liberar mais de duzentos 510(k)s por mês para que a indústria possa atender às necessidades do mercado de saúde nos EUA. Enquanto a representação gráfica (Ver Gráfico 1) indica a consistência da FDA em sua abordagem de revisão em meio à pandemia global. Embora o número não seja significativamente grande em comparação com o grande número de aplicações feitas globalmente. Espero que a FDA aumente seus recursos no futuro. Portanto, os fabricantes exigem uma preparação e uma apresentação cuidadosa para garantir a sua primeira tentativa.
| Ano de 2018 | Ano 2019 | Ano 2020 | |
| Total 510(k)s | 3025 | 2894 | 2901 |
| Total com resumos | 2885 | 2735 | 2759 |
| Total com declarações | 140 | 159 | 142 |
Tabela 1: A tabela indica o 510(k) anual liberado entre janeiro de 2018 - dezembro de 2020

Exigências da FDA 510k: Encontrando as chaves
A menos que bem versado com a FDA 510(k) preparação e apresentação pode ser realmente estressante e desafiador. Se um fabricante de dispositivos pretende comercializar um dispositivo nos EUA que seja das Classes I, II e III, mas não um isento de um 510(k) ou que faz não exigir um pedido de Aprovação Pré-Mercado (PMA) e, em seguida, sua elegibilidade para um 510(K). Prosseguindo, o fabricante deve ter uma clareza sobre os termos "dispositivo predicado" e "equivalência substancial (SE)". Um dispositivo legalmente comercializado nos EUA para o qual um fabricante deseja reivindicar uma equivalência é geralmente conhecido como um dispositivo qualificado. Por outro lado, equivalência substancial significa estabelecer uma comparação baseada em evidências para um novo dispositivo que um fabricante deseja colocar no mercado dos EUA é seguro e eficaz, já que o dispositivo qualificado existente é particularmente com relação ao mesmo uso pretendido, características tecnológicas. No caso do novo dispositivo diferir em características tecnológicas, ele não deve diferir em termos de segurança e eficácia e o fabricante é capaz de demonstrar que o dispositivo é tão seguro e eficaz quanto o dispositivo existente legalmente comercializado.
Desbloqueando o processo
Alguns dos fatores centrais para um 510(k) bem sucedido envolvem a classificação correta do código do produto, identificação e disponibilidade de um dispositivo predicado no mercado americano, comparação de equivalência substancial bem planejada com evidências, um sistema de gerenciamento de qualidade robusto, controles de projeto, aderência estrita aos formulários atuais da FDA e melhor utilização da lista de verificação de recusa de aceitação (RTA) (consulte também a figura 1). A lista de verificação de recusa de aceitação (RTA) orienta sobre os vários critérios de aceitação que a FDA segue enquanto realiza uma revisão substantiva de um 510(k). Para todos os três tipos de 510(k), há diferentes tipos de RTA disponíveis. A lista de verificação de RTA pode ser acessada a partir de aqui.
O fabricante deve observar que não é o número de páginas ou simplesmente apresentar os resultados dos testes ou estudos que lhes daria um 510(k) liberação uma escrita seqüencial bem explicada (como recomendado pela FDA em seu documento de orientação) baseada em justificativas e evidências científicas que serão cuidadosamente revisadas pela FDA dentro de um prazo fixo, acabando por atingir o 510(k). A maioria do 510(k) é rejeitada por falta de clareza ou provas inadequadas para reivindicar uma equivalência substancial [como no 510(k) tradicional] ou comparação insuficiente com as normas de conformidade reconhecidas pela FDA [como no 510(k) abreviado]. Observa-se que a falha no gerenciamento dos controles de projeto e a falta de um sistema de qualidade bem estabelecido é outra grande razão para tal falha. Finalmente, a indisponibilidade de uma equipe competente ou a falta de compreensão clara sobre a regulamentação é um fator inegável que contribui para a rejeição, além do acima mencionado.


Figura 1: Pontos-chave a considerar para uma Notificação Premarket 510(k)
Tipos de FDA 510(k) Notificação prévia ao mercado
Em geral, existem três tipos de 510(k)s que um fabricante pode apresentar ao FDA (veja figura 2). Eles são:
(1) Tradicional 510(k) - Os mais comuns e a maioria dos 510(k) estão neste tipo de aplicação,
(2) Especial 510(k) - Requerido somente quando são feitas alterações no rótulo(s) ou no projeto ou certas mudanças na indicação para uso em um dispositivo existente previamente liberado. O conteúdo deve atender aos requisitos definidos no 21 CFR Parte 807.87 e 21 Parte 807.90,
(3) Abreviado 510(k) - Aplica-se no caso de um fabricante ser capaz de produzir relatórios sobre o uso de controle ou orientação especial ou declaração de conformidade com base nas normas reconhecidas pela FDA.


Figura 2: Tipos de Notificação Premarket 510(k)
O processo de apresentação do 510k
Antes de uma apresentação, o fabricante terá que registrar sua organização junto à FDA. O processo é denominado registro de estabelecimento de acordo com o 21 CFR Parte 807 após o pagamento de taxas diretamente à FDA e o mesmo deverá ser renovado anualmente. Para o exercício financeiro atual de 2021, a taxa é de $ 5.546 para o registro de um estabelecimento. Uma vez, deve-se verificar as taxas exatas, visitando o site oficial do programa de taxas de usuários da FDA.
A FDA recomenda 20 seções em um 510(k) tradicional ou abreviado, mas não necessariamente todas as seções devem ser aplicáveis a um fabricante. Às vezes, se uma informação em uma determinada seção não se aplicar a seu dispositivo, eles podem incluir o título da seção e escrever "Esta seção não se aplica" ou "N/A" sob o mesmo. A seção principal recomendada do 510(k), conforme aconselhado no documento de orientação pela FDA, está listada abaixo:
- Folha de cobertura de taxa de usuário de dispositivo médico (Formulário FDA 3601): Indica o recebimento de uma taxa de usuário paga ao FDA pelo fabricante.
- Center for Devices and Radiological Health (CDRH) Premarket Review Submission Submission Cover Sheet (Form FDA 3514): Este é um formulário voluntário para fornecer todo tipo de informação administrativa à FDA sobre a organização e apresentação.
- A 510(k) Carta de Apresentação: Uma descrição sobre o propósito, conteúdo e informações administrativas sobre o 510(k) deve ser incorporada a esta carta. Recomenda-se consultar o Anexo A do "Formato para a Orientação Tradicional e Abreviada do 510(k)s para a Indústria e Funcionários da Administração de Alimentos e Drogas; dtd 13 de setembro de 2019".
- Indicação de uso (Formulário FDA 3881): Deve ser uniforme em todo o 510(k). Também deve definir se o dispositivo deve ser comercializado como prescrição ou uso no balcão (OTC).
- 510(k) Resumo ou 510(k) Declaração: A ser preparado de acordo com o 21 CFR parte 807. Aqui espera-se que o fabricante faça um resumo do 510(k) e incorpore informações sobre o restante do conteúdo.
- Declaração de Verdade e Precisão: É uma declaração de uma pessoa autorizada da organização certificando que todas as informações submetidas à FDA relacionadas ao 510(k) são verdadeiras e precisas.
- Resumo e Certificação Classe III: Aplicável somente a dispositivos de Classe III. É um resumo de segurança e eficácia e uma garantia de que uma busca razoável foi realizada e o fabricante tem todas as informações de segurança relevantes baseadas em dispositivos comercializados similares.
- Certificação Financeira ou Declaração de Divulgação: Se um fabricante apresenta provas clínicas, uma declaração de divulgação pelo investigador clínico deve ser anexada. O formulário 3454 ou o formulário 3455 da FDA pode ser encaminhado.
- Declarações de conformidade e relatórios resumidos: Aqui serão fornecidas informações relacionadas ao uso de normas de consenso voluntário ou a base de uso geral de tais normas.
- Descrição do dispositivo: Uma breve descrição do projeto do dispositivo, modelos ou acessórios deve ser incluída na seção.
- Resumo Executivo/Comparação de Predicados: Uma breve descrição do dispositivo, indicações de uso e tecnologia juntamente com a tabela de comparação de dispositivos é recomendada nesta seção.
- Discussão da Equivalência Substancial: Uma comparação detalhada entre o dispositivo do fabricante e o dispositivo predicado para demonstrar a equivalência substancial.
- Rotulagem proposta: Incluirá a rotulagem proposta para o dispositivo médico conforme 21 CFR 807.87(e) ou conforme 21 CFR 809.10 requisitos no caso de um dispositivo de diagnóstico in vitro.
- Esterilização e vida de prateleira: O método de esterilização, a validação relevante e o prazo de validade a ser incluído nesta seção.
- Biocompatibilidade: Protocolo de estudos, relatórios e garantia de que os estudos de biocompatibilidade foram realizados de acordo com as Boas Práticas de Laboratório. A FDA recomenda o uso da ISO 10993 para estudos de biocompatibilidade.
- Software: Se o dispositivo incorporar software, esta seção será aplicável.
- Compatibilidade Eletromagnética e Segurança Elétrica: Aplicável principalmente para dispositivos elétricos ou ativos. A FDA recomenda o uso do ANSI/AAMI (ES) 60601-1 para testes de segurança geral ou um método equivalente.
- Teste de desempenho - Bancada: Recomenda-se incluir os vários testes de desempenho realizados pelo fabricante ou em um laboratório de terceiros, que podem incluir, mas não se limitam aos resultados de testes mecânicos, de engenharia ou biológicos.
- Teste de desempenho - Animal: Se foram realizados estudos com animais e estão incluídos na apresentação, a FDA recomenda descrever os testes e fornecer os resultados que suportam as características de desempenho.
- Teste de desempenho - Clínico: Se o envio incluiu dados/estudos clínicos, a FDA espera a inclusão de informações sobre o protocolo e objetivo do estudo clínico, métodos de teste, pontos finais do estudo e ferramentas estatísticas utilizadas no estudo clínico.
Por favor, esteja ciente de que não há um modelo padrão ou tudo em um pronto para preencher o formato de aplicação 510(k); embora o 21 CFR Part 807, Subpart-E da FDA descreva o procedimento e oriente um fabricante sobre o registro do estabelecimento e a listagem de dispositivos. Além disso, vários formulários relevantes associados a tal apresentação, que são úteis para a preparação, podem ser baixados do link oficial da FDA, ou seja https://www.fda.gov/medical-devices/premarket-notification-510k/510k-forms . É aconselhável consultar as diversas orientações do FDA sobre o 510(k), disponíveis em domínio público, a fim de atender às exigências regulamentares e documentar em conformidade.Embora não seja uma prática realizar inspeções nas instalações de 510(k), mas o fabricante deve implementar um sistema de qualidade robusto, de acordo com a exigência do 21 CFR Parte 820 e estar pronto para a inspeção, caso ela ocorra.
Há também uma provisão conhecida como "Programa de Revisão por Terceiros" para certos dispositivos de baixo a moderado risco. Como o nome sugere, não é a FDA que revisa diretamente a aplicação 510(k), mas a organização terceira credenciada aprovada pela FDA faz o trabalho. Com base na revisão e recomendação feitas pelo terceiro credenciado, a FDA conclui uma decisão sobre uma autorização 510(k). Atualmente, existem 10 organizações terceirizadas disponíveis. Para um apresentante ou fabricante, é aconselhável verificar se seu dispositivo é elegível para uma análise de terceiros em https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfpcd/classification.cfm
510k Linha do tempo de aplicação
Um remetente ou fabricante ao submeter um 510(k) ao FDA também exige preparar uma cópia eletrônica de seu 510(k) e submeter o mesmo ao CDRH ou CBER no centro de controle de documentos (DCC). Com base nas informações apresentadas, o fabricante será solicitado a fornecer informações adicionais durante as diferentes etapas de revisão (Consulte a figura 3).
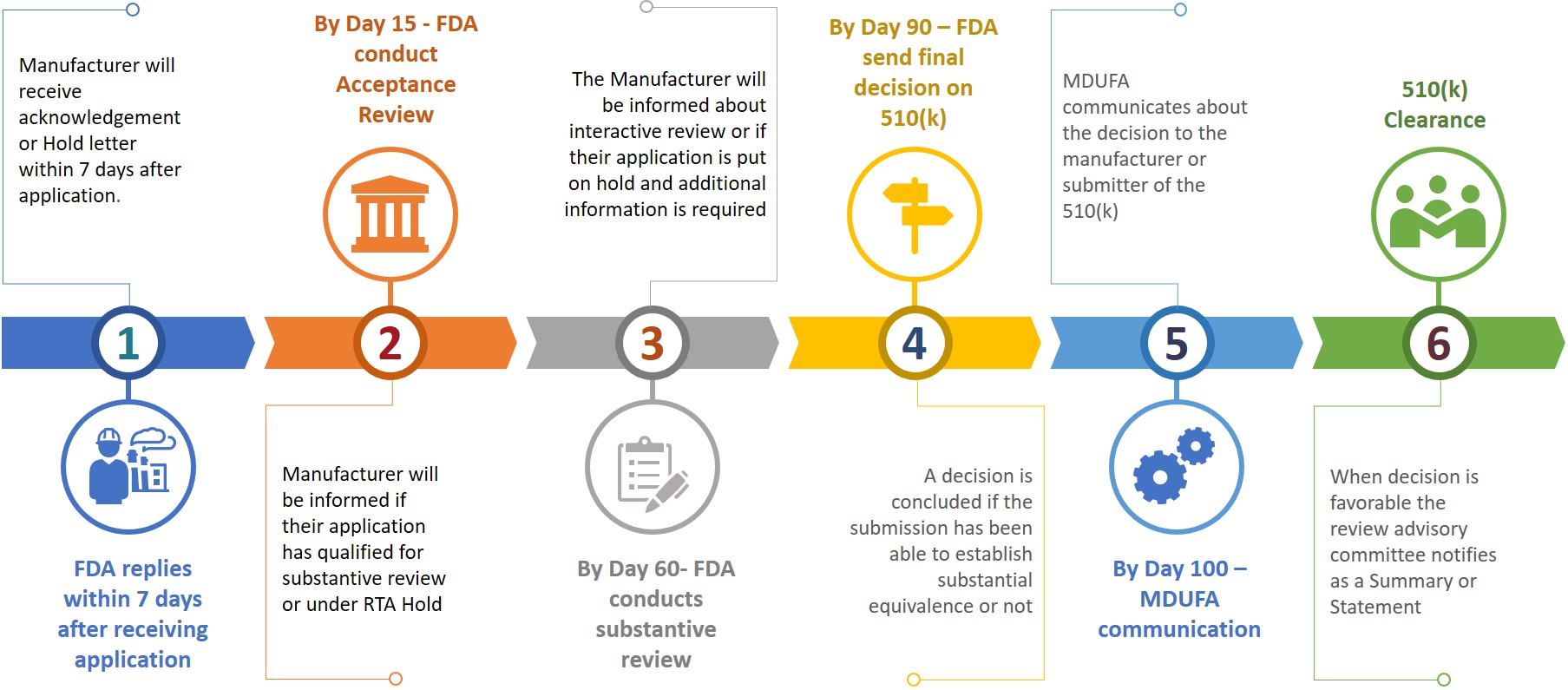

Figura 3: Linha do tempo de revisão tentativa
O apresentante terá 180 dias de calendário para responder quando receber um RTA ou se o FDA quiser informações adicionais. A não resolução de qualquer problema dentro de 18 dias corridos resultará na exclusão automática do sistema de revisão ou será considerada retirada. Em caso de exclusão ou retirada de um 510(k), o remetente terá que reaplicar um novo pedido após pagar a taxa necessária enquanto o número K pode ser citado no novo pedido para o mesmo dispositivo. Uma liberação do 510(k) pode ser obtida dentro de 100 dias após a apresentação, enquanto que pode levar de 6 a 9 meses para obter a liberação.
Referências
- O Programa 510(k): Evaluating Substantial Equivalence in Premarket Notifications [510(k)] Guidance for Industry and Food and Drug Administration Staff Document issued on: 28 de julho de 2014.
- Formato para Tradicional e Abreviado 510(k)s Guidance for Industry and Food and Drug Administration Staff; Documento emitido em 13 de setembro de 2019.
- [1]A visão geral da tecnologia médica (EUA), preparada em colaboração com a Unidade de Indústria e Análise (I&A) da Administração de Comércio Internacional https://www.selectusa.gov/medical-technology-industry-united-states (Último acesso em 11 de junho de 2021)


Sobre o autor
Sundeep Agarwal, Especialista no assunto e consultor da FDA, CE (MDR & IVDR)
With a decade of experience, he is globally sought-after Leader, Speaker & Consultant in the field of QA & RA, Quality Management System, Product Design & Development, Gerenciamento de risco, Commercial Scale-up, Industrial Manufacturing and Clinical Studies of medical devices.An active member of a Technical Group (Software as Medical Device) at Asian Harmonization Working Party.He joins Medical Device industry/government, collaborated conferences a speaker and panelist frequently on ISO 13485, EU MDR, IVDR, CE Certification, CER, PMS, USFDA, 510(K), ISO 14971, MDSAP, Combination Devices, Inteligência Artificial , etc. He prominently serves as a guest lecturer in various MBA and Pharmacy educational institutions in India. Entre em contato diretamente com ele para um projeto sobre Kolabtree.
Kolabtree helps businesses worldwide hire freelance scientists and industry experts on demand. Our freelancers have helped companies publish research papers, develop products, analyze data, and more. It only takes a minute to tell us what you need done and get quotes from experts for free.


Unlock Corporate Benefits
• Secure Payment Assistance
• Onboarding Support
• Dedicated Account Manager
Sign up with your professional email to avail special advances offered against purchase orders, seamless multi-channel payments, and extended support for agreements.