


Shreya Chenni, scrittore regolatore freelance for medical devices, provides a 10-minute guide to FDA design controls for your dispositivo medico.
Una delle cause principali dei richiami di dispositivi medici è la mancanza di controllo della progettazione come identificato dalla FDA [3,3a]. I controlli pre-produzione sono stati poi aggiunti ai regolamenti GMP dei dispositivi. I controlli di progettazione sono un insieme interconnesso di pratiche e procedure che sono incorporate nel processo di progettazione e sviluppo, cioè un sistema di controlli ed equilibri. I controlli di progettazione aumentano la probabilità che il progetto trasferito alla produzione si traduca in un dispositivo adeguato all'uso previsto.
Applicabilità
Tutti i dispositivi di classe II, III e i seguenti dispositivi di classe I sono soggetti a controlli di progettazione:
- Devices automated with computer software
- 868.6810 Catetere, aspirazione tracheobronchiale
- 878.4460 Guanto, da chirurgo
- 880.6760 Contenimento, protezione
- 892.5650 Sistema, applicatore, radionuclide, manuale
- 892.5740 Sorgente, teleterapia con radionuclidi
- Dispositivi automatizzati con software per computer
- Cateteri di aspirazione tracheobronchiali
- Surgeon’s gloves
- Vincoli di protezione
- Sistema, radionuclide, applicatore, manuale
- Sorgente, teleterapia con radionuclidi
Design controls apply to all D&D activities – for novel or improved devices being developed in the pre-market phase, as well as for changes to existing, marketed devices. Design controls do not apply to ricerca activities conducted during the proof of concept stage.
Design controls can be applied to any sviluppo del prodotto process. The following flowcharts are examples of the design controls applied to a traditional Waterfall design process and V-model process for Software (SW).
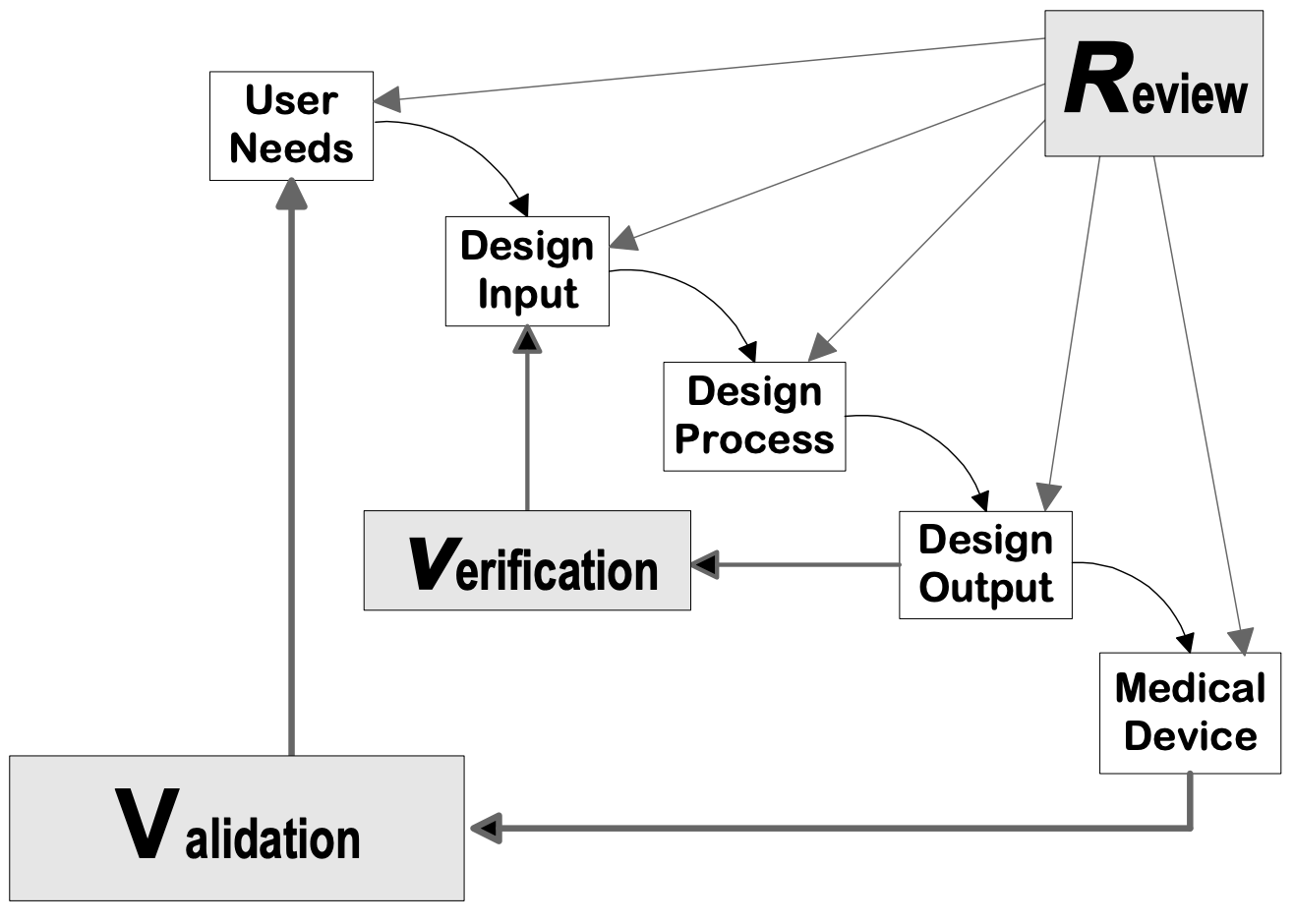

Fonte: Guida al controllo della progettazione per i produttori di dispositivi medici, CDRH-FDA
Fasi di progettazione del dispositivo
Fase di pianificazione di D&D
I piani di D&D dovrebbero essere stabiliti e mantenuti. Il piano deve descrivere o fare riferimento alle attività di progettazione e sviluppo e assegnare le responsabilità per l'attuazione. Come minimo l'FDA raccomanda di includere quanto segue nel piano:
- Scopi e obiettivi del programma D&D
- Delimitazione delle responsabilità delle attività di progettazione
- Identificare i compiti principali, i deliverable e assegnare la responsabilità per ogni compito
- Programmazione del compito principale in conformità con il calendario di sviluppo principale
- Identificare le principali revisioni e i punti di decisione
- Identificare i revisori, il team di revisione e le procedure che i revisori devono seguire
- Controlli della documentazione di progetto
- Attività di notifica
Il piano dovrebbe essere rivisto, aggiornato e approvato man mano che il progetto e lo sviluppo si evolvono.
Fase di ingresso del progetto
This is the starting point for product design. The medical device is designed and developed to meet the user requirements. Gather the user requirements from various sources such as customer surveys, feedback from the physicians, complaints. These requirements are transferred into design inputs.
- Ingressi di design: Questi sono i requisiti fisici e di prestazione di un dispositivo che sono usati per la progettazione del dispositivo. La fase di input della progettazione consiste nel convertire i requisiti dell'utente in requisiti del prodotto. I requisiti normativi sono considerati nella definizione degli input di progettazione. I requisiti di input di progettazione devono essere completi, non ambigui e verificabili oggettivamente. I requisiti di input possono essere raggruppati in 3 categorie:
- Requisiti funzionali, che descrivono cosa fa il dispositivo. Per esempio: La sedia a rotelle si sposta in avanti quando viene indicato dall'utente
- Requisiti di prestazione, che specificheranno quanto e come il dispositivo dovrebbe funzionare. Per esempio: La sedia a rotelle deve muoversi con una velocità di 2m/s in direzione di marcia
- Requisiti di interfaccia, specificano le caratteristiche del dispositivo che sono critiche per la compatibilità con i sistemi esterni, come l'interfaccia utente/paziente. Per esempio: La sedia a rotelle è dotata di pulsanti con simboli che indicano la direzione
- Ecco un esempio che definisce i bisogni degli utenti e li converte in input di progettazione:
- Necessità dell'utente
Il dispositivo deve essere portatile e abilitato al bluetooth
- Necessità dell'utente
-
- Ingresso al design
Identificare la FDA applicabile riconosciuta o uno standard internazionale a cui essere conformi. Come ad esempio:
-
-
- IEEE ANSI C63.27-2017 Standard nazionale americano per la valutazione della coesistenza wireless
- AAMI TIR 69: Association for the Advancement of Medical Instrumentation – Gestione del rischio of Radio-frequency Wireless Coexistence for Medical Devices and Systems (2017)
- IEC 60601-1-2 Edition 3: 2007: Medical Electrical Equipment – Part 1-2: General Requirements for Safety – Collateral Standard: Electromagnetic Compatibility – Requirements and Tests
- UL 2054 – Standard for Household and Commercial Batteries
-
Elenca le prestazioni e altri input specifici. Ad esempio:
-
-
- Il dispositivo deve essere alimentato in corrente continua
- Deve essere usato il modulo Bluetooth
- Peso: circa 6lbs o 6lbs+/- 2lbs (Gli input dovrebbero avere limiti quantitativi per garantire la verifica)
-
Fase di uscita del progetto
Gli output di progettazione sono i risultati di uno sforzo di progettazione in ogni fase di progettazione e alla fine dello sforzo totale di progettazione. Esempi di output di progettazione sono i disegni tecnici, l'etichettatura, le istruzioni di lavoro e altre specifiche del prodotto. Altri output di progettazione includono i risultati dell'analisi dei rischi, i risultati delle attività di verifica, i risultati dei test di biocompatibilità e il codice sorgente del software.
Gli output di progettazione non dovrebbero essere rilasciati prima della revisione e dell'approvazione da parte del personale responsabile. Si dovrebbe anche notare che qualsiasi modifica al dispositivo dopo l'approvazione degli output/input di progettazione sarà controllata attraverso la revisione e l'approvazione da parte del personale interessato. Una revisione del progetto è richiesta alla fine di questa fase.
Fase di revisione del progetto
Una revisione del progetto dovrebbe essere tenuta dopo la fase di uscita del progetto. Le revisioni del progetto dovrebbero seguire procedure stabilite ed essere documentate nel Design History File (DHF). I partecipanti a ogni revisione del progetto dovrebbero avere rappresentanti di tutti i gruppi funzionali.
Si raccomanda di condurre revisioni formali alla fine di importanti pietre miliari del progetto. Comunemente, le revisioni del progetto vengono condotte dopo la fase di uscita del progetto, la fase di V&V e la fase di trasferimento del progetto. Questo dipende anche dalla complessità dello sviluppo del dispositivo. La FDA richiede almeno una revisione del progetto.
Fase di verifica del progetto
La verifica del progetto è la conferma con prove oggettive che l'output del progetto soddisfa l'input del progetto. Fondamentalmente, è Design Input = Design Output. Le attività di verifica dovrebbero essere eseguite secondo le procedure stabilite. Esempi di verifica includono test EMC ed elettrici, ispezione visiva, attività di test non clinici, analisi ad albero dei guasti del processo o del progetto e analisi dei modi e degli effetti dei guasti. La verifica assicura che i requisiti tecnici delle specifiche del prodotto siano soddisfatti. Tutte le attività di verifica devono essere documentate.
Matrice di tracciabilità: Questo documento consiste in input e output di progetto elencati in un formato tabellare. Per ogni input si fa riferimento all'output corrispondente. Questo metodo di verifica è usato quando gli input e gli output sono entrambi documenti.
Fase di convalida del progetto
Convalida della progettazione significa stabilire con prove oggettive che le specifiche (requisiti specificati) sono conformi alle esigenze dell'utente e all'uso previsto.
- Convalida del processo significa stabilire con prove oggettive che un processo produce costantemente un risultato o un prodotto che soddisfa le sue specifiche predeterminate.
- Convalida della progettazione significa stabilire con prove oggettive che le specifiche del dispositivo sono conformi alle esigenze dell'utente e all'uso previsto.
La convalida è tipicamente condotto in condizioni reali o simulate. Examples of validation include studi clinicivalutazione clinica, test sui fattori umani, confezionamento ed etichettatura, analisi e ispezioni. I risultati delle attività di convalida e/o i rapporti di convalida devono essere documentati e faranno parte del Design History File (DHF).
Fase di trasferimento del progetto
Dopo il completamento della fase V&V, avviene il trasferimento del progetto. Questo include il trasferimento del design del dispositivo nelle specifiche del prodotto assicurando la qualità del dispositivo. Questa fase è molto critica perché una volta che la produzione del dispositivo inizia, sarà soggetta al controllo delle modifiche al progetto e può comportare una perdita finanziaria se si verificano problemi. Il trasferimento del design dovrebbe avvenire secondo la procedura stabilita. Inoltre, ci si dovrebbe assicurare che i documenti che includono le specifiche del prodotto siano rivisti e approvati prima di iniziare il trasferimento del progetto.
Fase delle modifiche al progetto
Il controllo delle modifiche al progetto inizia con il trasferimento del progetto e continua per tutto il ciclo di vita. Qualsiasi modifica al progetto dopo il trasferimento del progetto comporterà un Engineering Change Notice (ECN) da eseguire secondo una procedura stabilita. Bisogna assicurarsi che con ogni modifica al progetto, anche i documenti correlati come il rapporto sulla gestione dei rischi, le istruzioni per l'uso, i rapporti di verifica e di convalida devono essere rivisti e aggiornati.
Design History File (DHF)
Il DHF è specifico per la FDA statunitense. ISO 13486:2016 non richiede al produttore di mantenere un DHF.
Per ogni progetto viene mantenuto un Design History File che include tutti i risultati di ogni fase. Include le ultime informazioni sul prodotto. I documenti di progettazione e sviluppo devono essere facilmente disponibili e accessibili come e quando necessario. I contratti di progettazione e sviluppo dovrebbero specificare esplicitamente il diritto del produttore alle informazioni di progettazione e stabilire standard per la forma e il contenuto della documentazione di progettazione.
In pratica, i controlli di progettazione forniscono ai manager e ai progettisti una migliore visibilità del processo di progettazione. Con una migliore visibilità, i manager sono autorizzati a dirigere più efficacemente il processo di progettazione, cioè a riconoscere prima i problemi, a fare le correzioni e a regolare l'allocazione delle risorse. I progettisti beneficiano sia di una migliore comprensione del grado di conformità di un progetto ai bisogni degli utenti e dei pazienti, sia di una migliore comunicazione e coordinazione tra tutti i partecipanti al processo.
Hai bisogno di aiuto per capire e implementare i controlli di progettazione FDA per il tuo dispositivo medico? Contatta l'esperto consulenti di dispositivi medici su Kolabtree.
Riferimenti:
- FDA, guida al controllo della progettazione per i produttori di dispositivi medici
- Pratiche di regolamentazione medica, una prospettiva internazionale, Val Theisz
- Controllo del design, presentazione di Joseph Tartal
3a. Registro Federale / Vol. 61, No. 195
Kolabtree helps businesses worldwide hire freelance scientists and industry experts on demand. Our freelancers have helped companies publish research papers, develop products, analyze data, and more. It only takes a minute to tell us what you need done and get quotes from experts for free.


Unlock Corporate Benefits
• Secure Payment Assistance
• Onboarding Support
• Dedicated Account Manager
Sign up with your professional email to avail special advances offered against purchase orders, seamless multi-channel payments, and extended support for agreements.