

Nare Simonyan, freelance regulatory affairs specialist de Kolabtree, fournit un guide complet sur un dossier réglementaire et son format.
Introduction : Qu'est-ce qu'un dossier réglementaire ?
Le dossier réglementaire est un ensemble de documents pouvant inclure toutes les informations requises concernant des médicaments et/ou des génériques nouvellement développés, qui sont exigées par les autorités réglementaires de l'UE et des États-Unis pour l'octroi d'autorisations de mise sur le marché. Les principales informations incluses dans le dossier sont des informations administratives, des données relatives à la qualité, à la sécurité et à l'efficacité du produit pharmaceutique, qui peuvent être soumises au format CTD (Common Technical Document) en version papier ou électronique. Étant donné que les informations soumises en format papier étaient énormes, les agences encouragent désormais les demandes à être soumises en format eCTD.
CTD (Common Technical Document)
En 2003, les membres de l'ICH (International Council of Harmonization) ont convenu de rassembler toutes les informations relatives à la qualité, à la sécurité et à l'efficacité dans un format commun appelé CTD. Le CTD est un format/structure pour les modules 1 à 5 de la NDA (New Drug Application), de la MAA (Marketing Authorization Application) et des demandes médicinales globales. Le module 1 contient des informations administratives régionales qui sont différentes pour chaque pays. Les modules 2, 3, 4 et 5 sont communs à toutes les régions. La structure du CTD s'applique à la fois aux demandes de recherche et aux demandes commerciales (IND (Investigational New Drug) et NDA (US) ; IMPD (Investigational Medicinal Product Dossier) et MAA (EU), et les demandes globales).
The CTD was primarily used for new marketing applications such as NDA, BLA (Biologics License Application), MAA, NDS (New Drug Substance), JNDA (Japanese New Drug Application), etc. With additional guidance and guidelines from the FDA and EMA, the CTD is now required for all applications, including those for les essais cliniques—IMPD and INDs. All Drug Master Files (DMF) and Active Substance Master Files (ASMF) must follow the structure of the CTD. The process of submitting regulatory dossier is regulated by Code of Federal Regulations (CFR) (Law in US) and Directives (Law in EU).
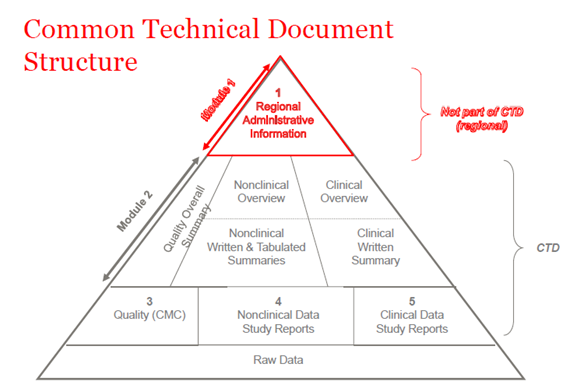

Figure 1. La structure du CTD
CTD-Module 1
Spécifications pour le module 1 américain : informations administratives et informations de prescription
La section Module 1 de l'e-CTD contient des documents administratifs et d'étiquetage. Toutes les demandes et soumissions connexes ont la même structure organisationnelle pour les documents du module 1.
Veuillez voir ci-dessous quelques spécifications sur les documents à fournir dans le cadre du module 1 :
Lettre de motivation (section 1.2)
Les lettres de motivation contiennent des informations pertinentes qui facilitent la communication dans le cadre du processus d'examen. Il est recommandé d'inclure les informations suivantes dans la lettre de motivation (voir "Document technique commun électronique (eCTD) v4.0 GUIDE DE CONFORMITÉ TECHNIQUE")
- Description réglementaire de la soumission, y compris les informations réglementaires appropriées, et tout hyperlien souhaité vers les informations soumises.
- Description technique de la soumission, y compris la taille approximative de la soumission (par exemple, 2 gigaoctets)
- Déclaration indiquant que la soumission est exempte de virus, avec une description du logiciel (nom, version et société) utilisé pour vérifier l'absence de virus dans les fichiers.
- Un point de contact réglementaire et technique pour la soumission, y compris l'adresse électronique.
Certification de la copie de terrain (section 1.3)
La certification de la copie de terrain doit être incluse dans l'eCTD pour les demandes de commercialisation. Il peut s'agir d'une lettre au bureau de district notifiant que la soumission eCTD sera soumise à la FDA. La lettre doit inclure :
- Numéro du médicament et de la demande
- Centre et division de l'Office des forêts
- La demande est en format eCTD.
Références (section 1.4)
Les références peuvent contenir les sous-sections suivantes (du contenu supplémentaire peut également être exigé)
- Lettres d'autorisation (LOA)
- Références croisées d'informations soumises antérieurement qui ne sont pas en format eCTD
Modifications de l'information (section 1.11)
Il peut être utilisé pour soumettre des réponses à des demandes d'information (DI), lorsque les informations soumises ne correspondent à aucune rubrique des modules 2, 3, 4 ou 5. Si la réponse à la demande d'information a une incidence sur les documents soumis dans les modules 2 à 5, les nouveaux documents ou les documents de remplacement doivent être soumis à l'emplacement approprié dans les modules 2 à 5 et référencés dans le document de la section 1.11.
Rapports annuels sur le marketing (section 1.13)
Pour chaque étude ou essai décrit dans les fichiers des exigences/engagements post-commercialisation, un signet doit être inclus.
Étiquetage (section 1.14)
La section 1.14 décrit comment fournir des documents d'étiquetage spécifiques. Il peut s'agir d'un projet d'étiquetage, d'un étiquetage final, d'un étiquetage de médicament listé, d'un étiquetage de médicament expérimental, d'un étiquetage étranger et d'un étiquetage de produit pour les soumissions 2253*. Les informations fournies peuvent contenir l'historique, le contenu et des échantillons d'étiquetage.
Formulaire FDA 2253* : Transmission des annonces et de l'étiquetage promotionnel des médicaments et des produits biologiques à usage humain
Publicités et matériel d'étiquetage promotionnel (section 1.15)
Les publicités et le matériel d'étiquetage promotionnel sont soumis à des restrictions aux États-Unis. Ils doivent être conformes aux exigences mentionnées dans le guide de la FDA. Fournir des soumissions réglementaires en format électronique et non électronique - Étiquetage promotionnel et matériel publicitaire pour les médicaments à prescription humaine.
Pour le matériel promotionnel soumis dans le cadre des exigences de rapport post-marketing, il est suggéré de fournir des liens hypertextes vers les références ou l'étiquetage. Les références améliorent l'efficacité d'un examen.
Stratégie d'évaluation et d'atténuation des risques (REMS) (section 1.16)
Le supplément REMS est destiné à proposer un nouveau REMS ou des modifications (majeures et/ou mineures) à un REMS approuvé. Il convient de sélectionner le type de REMS supplement dans le formulaire de soumission FDA approprié. Les évaluations de REMS, les révisions de REMS et les correspondances de REMS ne sont pas des suppléments. Dans ce cas type de soumission doit être sélectionné "autre" lorsque vous remplissez le formulaire FDA.
Spécifications pour le module 1 de l'Union européenne (UE) : Informations administratives et informations relatives à la prescription
Lettre de présentation (section 1.0)
Une lettre de présentation doit être fournie pour chaque demande. Les documents "Notes aux évaluateurs" peuvent être inclus en annexe de la lettre d'accompagnement, au cas où des informations supplémentaires doivent être fournies afin de faciliter la navigation.
Table des matières exhaustive (section 1.1)
Une table des matières complète doit être fournie pour chaque type de demande, qui peut contenir toutes les sections du module qui ont été soumises dans le cadre de la demande concernée. Dans le cas de nouvelles demandes, toutes les sections doivent être abordées.
Formulaire de demande (section 1.2)
Selon le type de soumission, le formulaire de demande correspondant doit être inclus dans le dossier réglementaire.
Les différents formulaires de demande sont disponibles sur le site web de la Commission européenne /
DG Enterprise :
- Nouvelles applications et applications d'extension http://ec.europa.eu/enterprise/pharmaceuticals/eudralex/homev2.htm#2b
- Applications des variations
http://ec.europa.eu/enterprise/pharmaceuticals/eudralex/homev2.htm#2c
- Demandes de renouvellement
http://ec.europa.eu/enterprise/pharmaceuticals/eudralex/homev2.htm#2c
Informations sur le produit (section 1.3)
La section 1.3 contient des informations concernant
- Résumé des caractéristiques du produit (RCP), étiquetage et notice (PIL) (Section 1.3.1)
- Maquette (Section 1.3.2)
- Echantillon (Section 1.3.3)
- Consultation des groupes de patients cibles (Section 1.3.4)
- Informations sur le produit déjà approuvées dans les États membres (Section 1.3.5) (le cas échéant)
- Braille (Section 1.3.6)
Informations sur les experts (section 1.4)
Dans cette section, il est nécessaire d'inclure des informations concernant les experts qui fournissent des rapports détaillés des documents et des informations qui constituent les modules 3, 4 et 5 conformément à l'article 12 de la directive 2001/83/CE. Pour une partie supplémentaire de cette section peut être utilisé un rapport d'expert signé pour les différentes parties scientifiques du dossier. Les exigences relatives aux rapports d'experts signés sont présentées ci-dessous :
- Le résumé global de la qualité, la vue d'ensemble / le résumé non clinique et la vue d'ensemble / le résumé clinique du module 2,
- Une déclaration signée par les experts du module 1.4.
- Une brève information sur le parcours scolaire, la formation et l'expérience professionnelle dans le module 1.4.
La ou les déclarations d'experts pertinentes doivent être fournies pour les demandes post-autorisation.
Dans les cas où les titulaires d'une autorisation de mise sur le marché souhaitent distinguer cette déclaration de tout autre type de déclaration.
les déclarations antérieures, le numéro de procédure pertinent de l'État membre/EMEA de référence.
peut être incluse sur le dessus.
Les sous-sections de l'information sur les experts sont présentées ci-dessous :
- Qualité (section 1.4.1)
- Non clinique (1.4.2)
- Clinique (section 1.4.3)
Pour de plus amples informations et des modèles pertinents, veuillez vous référer à l'article 12 et conformément à l'annexe I, partie I 1.4 de la directive 2001/83/CE.
Exigences spécifiques pour différents types d'applications (section 1.5)
Informations pour les applications bibliographiques (section 1.5.1)
Sur la base de l'article 10a de la directive 2001/83/CE, les demandeurs doivent fournir un document concis, résumant les raisons et les preuves utilisées pour démontrer que le ou les composants du médicament ont un usage bien établi, en tenant compte du niveau acceptable de sécurité et d'efficacité, comme indiqué dans la partie II.1 de l'annexe I de la directive 2001/83/CE.
Informations relatives aux demandes de médicaments génériques, "hybrides" ou biosimilaires (section 1.5.2)
Sur la base de l'article 10(1), 10(3) ou 10(4) de la directive 2001/83/CE, les demandeurs doivent fournir ici un document concis, résumant les raisons et les preuves utilisées pour démontrer le type de médicament pour lequel une demande est soumise. Les médicaments peuvent être des génériques, des produits hybrides et des biosimilaires.
Exclusivité (étendue) des données / du marché (section 1.5.3)
La section 1.5.3 est requise lorsque le titulaire de l'AMM/demandeur souhaite revendiquer l'exclusivité des données/du marché (supplémentaire) tout en demandant une nouvelle indication ou un changement de classification. Dans ce cas, les dispositions et exigences légales pertinentes doivent être prises en compte.
Circonstances exceptionnelles (section 1.5.4)
Selon l'article 22 de la directive 2001/83/CE et l'article 14, paragraphe 7, du règlement (CE) n° 726/2004, une autorisation peut être accordée en cas de circonstances exceptionnelles, lorsque des procédures spécifiques, concernant notamment la sécurité du médicament, la notification aux autorités compétentes de tout incident lié à son utilisation sont mises en place par le demandeur. Une autorisation ne peut être accordée que pour des exceptions objectives et vérifiables.
Autorisation conditionnelle de mise sur le marché (section 1.5.5)
La section mentionnée est applicable à la procédure centralisée. Les références de cette section sont l'article 14, paragraphe 7, du règlement (CE) n° 726/2004 et "...".Directive relative à l'application scientifique et aux modalités pratiques de l'autorisation de mise sur le marché conditionnelle".
Évaluation des risques environnementaux (section 1.6)
Conformément à l'article 8 (ca) et (g) de la directive 2001/83/CE, tout risque potentiel du médicament pour l'environnement doit être pris en compte par le demandeur lors de la demande d'autorisation de mise sur le marché. Les exigences de la directive concernent l'utilisation, le stockage et l'élimination des médicaments et ne sont pas applicables à la synthèse ou à la fabrication du produit. Les demandes d'autorisation de mise sur le marché de médicaments ne contenant pas d'OGM (section 1.6.1 "Non-OGM") et contenant des OGM (section 1.6.2 "OGM") doivent inclure une indication des risques potentiels dans le module 1 du dossier réglementaire.
Pour plus d'informations, veuillez vous référer à "Ligne directrice sur l'évaluation des risques environnementaux des médicaments à usage humain"
Informations relatives à l'exclusivité du marché des médicaments orphelins (section 1.7)
Cette section n'est applicable que pour les médicaments orphelins. Les informations requises sur les détails et la procédure sont présentes dans "Ligne directrice de la Commission européenne sur les aspects de l'application de l'article 8 du règlement (CE) n° 141/2000 : Évaluation de la similarité et/ou de la supériorité clinique des médicaments orphelins lors de l'évaluation des demandes d'autorisation de mise sur le marché et des modifications."
Les sections 1.7.1 Similitude et 1.7.2 Exclusivité de marché sont applicables, si le médicament orphelin a été autorisé pour la condition qui couvre l'indication thérapeutique proposée faisant l'objet de la demande, et qu'une période d'exclusivité de marché est en vigueur, les demandeurs doivent fournir un rapport critique traitant de la similitude éventuelle avec le médicament orphelin autorisé et concluant à la similitude ou à la "non" similitude (1.7.1). Si le médicament faisant l'objet de la demande d'autorisation de mise sur le marché est considéré comme "similaire" à un médicament orphelin couvert par les dispositions d'exclusivité commerciale susmentionnées, le demandeur doit en outre justifier l'une des dérogations prévues à l'article 8, paragraphe 3, points a) à c), du règlement (CE) n° 141/2000 (1.7.2).
Information relating to Pharmacovigilance (Section 1.8)
Système de pharmacovigilance (section 1.8.1)
La pharmacovigilance est la science et les activités liées à la détection, l'évaluation, la compréhension et la prévention des effets indésirables ou de tout autre problème lié aux médicaments.
Cela s'applique tout au long du cycle de vie du médicament, aussi bien au stade de la pré-approbation qu'à celui de la post-approbation. Le système de pharmacovigilance est un volet très important pour la demande d'autorisation de mise sur le marché.
Une description détaillée du système de pharmacovigilance doit être fournie conformément à l'article 8 (ia) de la directive 2001/83/CE. La description doit inclure la preuve que le demandeur dispose des services d'une personne qualifiée responsable de la pharmacovigilance.
La description du système de pharmacovigilance du titulaire de l'autorisation de mise sur le marché doit respecter les exigences et le format détaillés dans le volume 9A d'EudraLex.
Système de gestion des risques (section 1.8.2)
Detailed description of risk-management system must be provided according to Article 8 (ia) of Directive 2001/83/EC. The detailed description of a risk management system should be provided in the form of an EU Risk Management Plan (EU-RMP), as outlined in Volume 9A of EudraLex.
Informations relatives aux essais cliniques (section 1.9)
La section 1.9 doit être préparée conformément à l'article 8 (ib) de la directive 2001/83/CE pour qu'un rapport indiquant que les essais cliniques réalisés en dehors de l'Union européenne répondent aux exigences éthiques de la directive 2001/20/CE soit fourni, le cas échéant.
Cette section doit être fournie pour toutes les nouvelles demandes (y compris les demandes d'extension), et autres procédures réglementaires post-autorisation pertinentes (par exemple, les variations) pour lesquelles l'essai clinique a soumis des rapports.
Informations relatives à la pédiatrie (section 1.10)
Conformément aux articles 7, 8 et 30 du règlement (CE) n° 1901/2006 ("règlement pédiatrique") et à l'article 23 du règlement (CE) n° 1901/2006 ("règlement pédiatrique"), cette section est requise :
- Pour toute nouvelle demande*, pour un médicament qui n'est pas autorisé dans l'EEE
- Pour les demandes* de nouvelles indications, de nouvelles formes pharmaceutiques et de nouvelles voies d'administration, pour les médicaments autorisés qui sont protégés soit par un certificat complémentaire de protection, soit par un brevet permettant l'octroi d'un tel certificat.
- Pour les demandes d'autorisation de mise sur le marché à usage pédiatrique (PUMA)
*à l'exception des applications génériques, hybrides, biosimilaires et à usage bien établi et des médicaments traditionnels à base de plantes ou homéopathiques.
Pour le module 1 peuvent être fournis :
Réponses aux questions dans les cas où il est conseillé aux candidats d'inclure dans cette section un document qui énumère les questions avec le texte de réponse correspondant à chaque question, et lorsque les réponses contiennent également des données/documents nouveaux ou mis à jour concernant les modules 3, 4 et/ou 5. Ces données/documents doivent être placés dans les sections correspondantes de ces modules.
Données supplémentaires. Cette section est requise en fonction de la procédure d'autorisation. Des données supplémentaires peuvent devoir être fournies dans le cadre d'une demande de reconnaissance nationale, décentralisée ou mutuelle. Si ces données concernent les modules 2, 3, 4 et/ou 5, les documents doivent également être placés dans les sections correspondantes de ces modules. Les exigences spécifiques des États membres en matière de données supplémentaires peuvent être consultées sur le site Internet de la Commission européenne. Commission européenne.
CTD-Module 2
L'introduction générale au médicament est fournie dans la section Module 2 du dossier CTD, qui est harmonisé pour toutes les régions (le Conseil international d'harmonisation des exigences techniques relatives aux produits pharmaceutiques à usage humain (ICH)). Ce module est présenté par des documents de synthèse pour chacun des modules à venir : données sur la qualité, rapports d'études non cliniques et cliniques.
CTD-Module 3
Module 3 was identified by ICH as the Quality Module. Thus, “Quality” became the global term for CMC (chimie, manufacturing and controls). Il ne faut pas confondre les CMC avec les éléments du contrôle de la qualité, de l'assurance de la qualité, les SOP (procédures opératoires standard), les documents internes de l'entreprise (spécifications, dossiers de lot, etc.).
La section du module 3 est également harmonisée pour toutes les régions et fournit des informations chimiques, pharmaceutiques et biologiques sur les substances actives chimiques et les médicaments biologiques.
Tout au long du développement et après l'approbation d'un produit par les autorités sanitaires, les aspects liés à la chimie, à la fabrication et au contrôle (CMC) continuent d'évoluer et de changer.
Qualité pharmaceutique = CMC
CHIMIE
Découverte de nouvelles entités chimiques/molécules, purification des principes actifs (substance médicamenteuse), tests analytiques des matières premières et des produits finis.
MANUFACTURIERS
Fabrication à l'échelle du laboratoire de substances et de produits pharmaceutiques, fabrication de fournitures cliniques pour les études cliniques, mise à l'échelle jusqu'à la taille du lot commercial, produit commercial.
ET CONTRÔLES
Développer des spécifications/contrôles appropriés pour la substance médicamenteuse et le produit pharmaceutique afin de garantir la sécurité, l'efficacité et la qualité.
Toutes les régions exigent un dossier CMC/Qualité pharmaceutique
- Lancer et mener des essais cliniques,
- Pour soumettre un nouveau dossier d'autorisation de mise sur le marché (MAA, NDA, BLA, etc.)
- Répondre aux exigences réglementaires pour la gestion du cycle de vie et les changements post-approbation du produit.
Ce module comprend l'ensemble des documents relatifs à la substance médicamenteuse (DS) et au produit médicamenteux (DP).
Substance médicamenteuse (DS)
La section sur la substance médicamenteuse est présentée par sa description, y compris ses caractéristiques physiques, chimiques ou biologiques, le nom et l'adresse du fabricant de DS, les limites acceptables et les méthodes d'analyse utilisées pour assurer l'identité, la force, la qualité et la pureté de la substance médicamenteuse.
Des informations à l'appui de la stabilité de la substance médicamenteuse pendant les études toxicologiques et l'étude clinique proposée sont également incluses.
PRODUIT PHARMACEUTIQUE (PDP)
Une liste de tous les composants, qui peuvent inclure des alternatives raisonnables pour les composés inactifs, utilisés dans la fabrication du produit pharmaceutique, y compris les composants destinés à apparaître dans le produit pharmaceutique et ceux qui peuvent ne pas apparaître, mais qui sont utilisés dans le processus de fabrication sont décrits dans cette section.
Vous trouverez ci-dessous une liste des informations clés du produit pharmaceutique qui doivent être incluses dans le dossier réglementaire :
- La composition quantitative du produit pharmaceutique (expérimental ou commercial), y compris toute variation raisonnable.
- Le nom et l'adresse du fabricant du produit pharmaceutique.
- Description des procédures de fabrication et de conditionnement.
- Les limites acceptables et les méthodes analytiques utilisées pour garantir l'identité, la force, la qualité et la pureté du produit pharmaceutique.
CTD-Module 4
Rapports d'études non-cliniques
La section pertinente l'emplacement approprié pour les données individuelles sur les animaux est dans le rapport d'étude dans le document technique commun pour les applications qui seront soumises aux autorités réglementaires. Le module 4 est harmonisé pour les États-Unis et l'Union européenne sur la base des principes de la CIH, et contient toutes les sections et sous-sections nécessaires pour les rapports d'étude. Références aux lignes directrices de l'ICH Document technique commun (CTD).
CTD-Module 5
Rapports d'études cliniques
La section du module 5 concerne la structure et le contenu des rapports d'études cliniques. Cette partie du CTD présente les rapports d'études humaines/cliniques, les autres données cliniques et les références dans un Document Technique Commun (CTD) pour l'enregistrement d'un produit pharmaceutique à usage humain. Ces éléments doivent faciliter la préparation et l'examen d'une demande de commercialisation. Le module 5 est harmonisé pour les États-Unis et l'Union européenne sur la base des principes de la CIH, et contient toutes les sections et sous-sections nécessaires pour les rapports d'étude. Références aux lignes directrices de l'UE Document technique commun (CTD).
Besoin d'aide pour préparer un dossier réglementaire ? Voir et consulter rédacteurs réglementaires indépendants sur Kolabtree.
Références supplémentaires
- https://www.ich.org/page/ctd
- https://www.fda.gov/media/128163/download
- https://www.fda.gov/media/135573/download
- https://ec.europa.eu/health/sites/default/files/files/eudralex/vol-2/b/update_200805/ctd_05-2008_en.pdf
- https://ec.europa.eu/health/sites/default/files/files/eudralex/vol-1/dir_2001_83_consol_2012/dir_2001_83_cons_2012_en.pdf
- https://eur-lex.europa.eu/legal-content/EN/TXT/PDF/?uri=CELEX:02001L0083-20121116&from=DE
- https://eur-lex.europa.eu/legal-content/EN/TXT/PDF/?uri=CELEX:02004R0726-20130605&from=EN
- https://ec.europa.eu/health/sites/default/files/files/eudralex/vol-1/reg_2000_141_cons-2009-07/reg_2000_141_cons-2009-07_en.pdf
- https://www.ema.europa.eu/en/human-regulatory/research-development/clinical-trials-human-medicines
- https://eur-lex.europa.eu/legal-content/EN/TXT/?uri=celex%3A32006R1901
- https://clinicaltrials.gov/ct2/home
Kolabtree helps businesses worldwide hire freelance scientists and industry experts on demand. Our freelancers have helped companies publish research papers, develop products, analyze data, and more. It only takes a minute to tell us what you need done and get quotes from experts for free.


Unlock Corporate Benefits
• Secure Payment Assistance
• Onboarding Support
• Dedicated Account Manager
Sign up with your professional email to avail special advances offered against purchase orders, seamless multi-channel payments, and extended support for agreements.