

Dr. Javier Quilez Oliete, ein erfahrener freelance bioinformatics consultant on Kolabtree, provides a comprehensive guide to DNA sequencing Datenanalyse, including tools and software used to read data.
Einführung
Die Desoxyribonukleinsäure (DNA) ist das Molekül, das den größten Teil der genetischen Information trägt eines Organismus. (Bei einigen Virustypen wird die genetische Information durch Ribonukleinsäure (RNA) übertragen). Nukleotide (üblicherweise durch die Buchstaben A, C, G oder T dargestellt) sind die Grundeinheiten von DNA-Molekülen. Konzeptionell, DNA-Sequenzierung ist der Prozess des Lesens der Nukleotide, aus denen ein DNA-Molekül besteht (z. B. "GCAAACCAAT" ist eine DNA-Kette mit 10 Nukleotiden). Aktuelle Sequenzierungstechnologien erzeugen Millionen solcher DNA-Leseabschnitte in einer angemessenen Zeit und zu relativ geringen Kosten. Zum Vergleich: Die Kosten für die Sequenzierung eines menschlichen Genoms - ein Genom ist der vollständige Satz von DNA-Molekülen in einem Organismus - haben sich auf $100 Barriere und kann innerhalb weniger Tage durchgeführt werden. Dies steht im Gegensatz zu der ersten Initiative zur Sequenzierung der menschliches Genomdas in einem Jahrzehnt fertiggestellt wurde und etwa $2,7 Milliarden kostete.
This capability to sequence DNA at high throughput and low cost has enabled the development of a growing number of sequencing-based methods and applications. For example, sequencing entire genomes or their protein-coding regions (two approaches known respectively as whole genome and exome sequencing) in disease and healthy individuals can hint to disease-causing DNA alterations. Also, the sequencing of the RNA that is transcribed from DNA—a technique known as RNA-sequencing—is used to quantify gene activity and how this changes in different conditions (e.g. untreated versus treatment). On the other side, chromosome conformation capture sequencing methods detect interactions between nearby DNA molecules and thus help to determine the spatial distribution of chromosomes within the cell.
Diesen und anderen Anwendungen der DNA-Sequenzierung ist gemeinsam, dass sie Datensätze in der Größenordnung von Gigabytes erzeugen, die Millionen von Lesesequenzen umfassen. Daher erfordert die Auswertung von Hochdurchsatz-Sequenzierungsexperimenten (HTS) umfangreiche Datenanalysefähigkeiten. Glücklicherweise gibt es für die meisten HTS-Datentypen spezielle Berechnungs- und Statistik-Tools und relativ standardisierte Analyse-Workflows. Während einige der (anfänglichen) Analyseschritte für die meisten Sequenzierungsdatentypen gleich sind, hängt die weitere nachgelagerte Analyse von der Art der Daten und/oder dem letztendlichen Ziel der Analyse ab. Im Folgenden gebe ich eine Einführung in die grundlegenden Schritte der Analyse von HTS-Daten und verweise auf gängige Tools.
Einige der folgenden Abschnitte befassen sich mit der Analyse von Daten, die mit Short-Read-Sequenzierungstechnologien erzeugt wurden (meist Illumina), da diese in der Vergangenheit den HTS-Markt dominiert haben. Neuere Technologien, die längere Lesezeiten erzeugen (z. B. Oxford-Nanopore-Technologien, PacBio) sind schnell auf dem Vormarsch. Da die Long-Read-Sequenzierung einige Besonderheiten aufweist (z. B. höhere Fehlerquoten), werden derzeit spezielle Werkzeuge für die Analyse dieser Art von Daten entwickelt.
Qualitätskontrolle (QC) von Rohdaten
Der eifrige Analytiker beginnt die Analyse mit FASTQ-Dateien; der FASTQ-Format ist seit langem der Standard für die Speicherung von Short-read-Sequenzierungsdaten. Im Wesentlichen enthalten FASTQ-Dateien die Nukleotidsequenz und die basenbezogene Aufrufqualität für Millionen von Reads. Obwohl die Dateigröße von der tatsächlichen Anzahl der Reads abhängt, sind FASTQ-Dateien in der Regel groß (in der Größenordnung von Megabytes und Gigabytes) und komprimiert. Die meisten Tools, die FASTQ-Dateien als Eingabe verwenden, können sie in komprimiertem Format verarbeiten. Um Speicherplatz zu sparen, wird empfohlen, sie nicht zu dekomprimieren. Als Konvention werde ich hier eine FASTQ-Datei mit einer Sequenzierungsprobe gleichsetzen.
FastQC ist wahrscheinlich das beliebteste Tool zur Durchführung der Qualitätskontrolle von Rohdaten. Es kann über eine visuelle Schnittstelle oder programmatisch ausgeführt werden. Während die erste Option für Benutzer, die sich mit der Befehlszeilenumgebung nicht wohlfühlen, bequemer ist, bietet die letztere eine unvergleichliche Skalierbarkeit und Reproduzierbarkeit (man denke nur daran, wie mühsam und fehleranfällig es sein kann, das Tool manuell für Dutzende von Dateien auszuführen). Wie auch immer, die Hauptausgabe von FastQC ist eine HTML-Datei reporting key summary Statistik about the overall quality of the raw sequencing reads from a given sample. Inspecting tens of FastQC reports one by one is tedious and it complicates the comparison across samples. Therefore, you may want to use MultiQCdas die HTML-Berichte von FastQC (sowie von anderen nachgeschalteten Tools, z. B. Adapter Trimming, Alignment) in einem einzigen Bericht zusammenfasst.

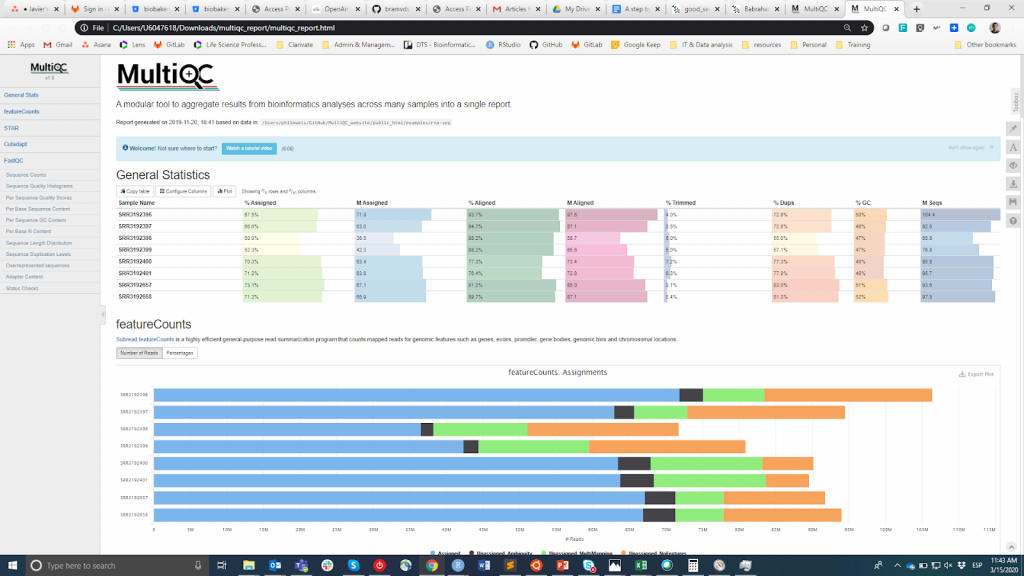
MultiQC
Die QC-Informationen sollen es dem Benutzer ermöglichen zu beurteilen, ob die Proben von guter Qualität sind und daher für die nachfolgenden Schritte verwendet werden können oder ob sie verworfen werden müssen. Leider gibt es keinen einheitlichen Schwellenwert auf der Grundlage der FastQC-Metriken, um Proben als von guter oder schlechter Qualität zu klassifizieren. Der von mir verwendete Ansatz ist der folgende. Ich erwarte, dass alle Proben, die dasselbe Verfahren durchlaufen haben (z. B. DNA-Extraktion, Bibliotheksvorbereitung), ähnliche Qualitätsstatistiken und eine Mehrheit von "Pass"-Flags aufweisen. Wenn einige Proben eine unterdurchschnittliche Qualität aufweisen, verwende ich sie dennoch in der nachgelagerten Analyse, wobei ich dies berücksichtige. Andererseits, wenn alle Proben im Experiment systematisch "Warnung" oder "Nicht bestanden" in mehreren Metriken erhalten (siehe dieses Beispiel), vermute ich, dass bei dem Experiment etwas schief gelaufen ist (z. B. schlechte DNA-Qualität, Bibliotheksvorbereitung usw.), und ich empfehle, es zu wiederholen.
Beschneiden lesen
Die Qualitätskontrolle von Rohdaten hilft, problematische Proben zu identifizieren, verbessert aber nicht die tatsächliche Qualität der Daten. Zu diesem Zweck müssen wir die Reads trimmen, um technische Sequenzen und qualitativ schlechte Enden zu entfernen.
Technische Sequenzen sind Überbleibsel des experimentellen Verfahrens (z. B. Sequenzieradapter). Wenn solche Sequenzen an die tatsächliche Sequenz des Reads angrenzen, kann das Alignment (siehe unten) die Reads an der falschen Position im Genom zuordnen oder das Vertrauen in ein bestimmtes Alignment verringern. Neben technischen Sequenzen können auch Sequenzen biologischen Ursprungs entfernt werden, wenn diese in den Reads stark vertreten sind. So kann beispielsweise bei suboptimalen DNA-Präparationsverfahren ein hoher Anteil an DNA-umgewandelter ribosomaler RNA (rRNA) in der Probe zurückbleiben. Sofern diese Art von Nukleinsäure nicht das Ziel des Sequenzierungsexperiments ist, erhöht die Beibehaltung von Reads, die von rRNA stammen, nur den Rechenaufwand der nachgeschalteten Schritte und kann die Ergebnisse verfälschen. Wenn der Anteil an technischen Sequenzen, rRNA oder anderen Verunreinigungen sehr hoch ist, was wahrscheinlich bereits bei der Qualitätskontrolle festgestellt wurde, sollten Sie die gesamte Sequenzierprobe verwerfen.
Bei der Short-Read-Sequenzierung wird die DNA-Sequenz Nukleotid für Nukleotid bestimmt (technisch gesehen ein Nukleotid pro Sequenzierzyklus). Mit anderen Worten: Die Anzahl der Sequenzierzyklen bestimmt die Leselänge. Ein bekanntes Problem der HTS-Sequenzierungsmethoden ist die Abnahme der Genauigkeit, mit der die Nukleotide bestimmt werden, wenn die Sequenzierungszyklen zunehmen. Dies spiegelt sich in einer allgemeinen Abnahme der Qualität des Callings pro Base wider, insbesondere gegen Ende des Reads. Wie bei technischen Sequenzen kann der Versuch, Reads mit minderwertigen Enden zu alignieren, zu Fehlplatzierungen oder schlechter Mapping-Qualität führen.
Zur Entfernung von technischen/verunreinigenden Sequenzen und minderwertigen Enden lesen Sie Trimming-Tools wie Trimmomatic und Cutadapt existieren und sind weit verbreitet. Im Wesentlichen entfernen solche Tools technische Sequenzen (intern verfügbar und/oder vom Nutzer bereitgestellt) und trimmen Reads auf der Grundlage der Qualität bei gleichzeitiger Maximierung der Leselänge. Reads, die nach dem Trimming zu kurz sind, werden verworfen (zu kurze Reads, z. B. <36 Nukleotide, erschweren den Alignment-Schritt, da sie wahrscheinlich mehreren Stellen im Genom zugeordnet sind). Sie sollten sich den Prozentsatz der Reads ansehen, die das Trimming überleben, da ein hoher Anteil an verworfenen Reads wahrscheinlich ein Zeichen für schlechte Datenqualität ist.
Abschließend führe ich in der Regel FastQC erneut auf den getrimmten Reads aus, um zu überprüfen, ob dieser Schritt effektiv war und die QC-Metriken systematisch verbessert hat.
Ausrichtung
Mit Ausnahmen (z.B. De-novo-Versammlung) ist das Alignment (auch als Mapping bezeichnet) bei den meisten HTS-Datentypen und -Anwendungen der nächste Schritt. Das Read-Alignment besteht in der Bestimmung der Position im Genom, von der die Sequenz des Reads stammt (in der Regel ausgedrückt als Chromosom:Start-Ende). Daher benötigen wir in diesem Schritt eine Referenzsequenz, an der wir die Reads ausrichten/zuordnen können.
Die Wahl der Referenzsequenz wird durch mehrere Faktoren bestimmt. Zum einen von der Art, von der die sequenzierte DNA stammt. Während die Zahl der Arten, für die eine qualitativ hochwertige Referenzsequenz zur Verfügung steht, zunimmt, kann dies bei einigen weniger erforschten Organismen noch nicht der Fall sein. In diesen Fällen sollten Sie die Reads an einer evolutiv nahen Art ausrichten, für die ein Referenzgenom verfügbar ist. Da es zum Beispiel keine Referenzsequenz für das Genom des Kojoten gibt, können wir für das Read-Alignment die Sequenz des nahe verwandten Hundes verwenden. Ebenso kann es sein, dass wir unsere Reads an einer eng verwandten Art ausrichten wollen, für die eine qualitativ hochwertigere Referenzsequenz existiert. Das Genom des Gibbons zum Beispiel ist zwar bereits veröffentlichtDieses ist in Tausende von Fragmenten zerlegt, die die Organisation dieses Genoms in Dutzende von Chromosomen nicht vollständig wiedergeben. In diesem Fall kann es von Vorteil sein, den Abgleich anhand der menschlichen Referenzsequenz durchzuführen.
Ein weiterer zu berücksichtigender Faktor ist die Version der Referenzsequenz, da neue Versionen veröffentlicht werden, wenn die Sequenz aktualisiert und verbessert wird. Wichtig ist, dass die Koordinaten eines bestimmten Alignments zwischen den Versionen variieren können. So finden sich beispielsweise mehrere Versionen des menschlichen Genoms in der UCSC Genom-Browser. Auf jeden Fall empfehle ich, auf die neueste Assembly-Version zu migrieren, sobald diese vollständig freigegeben ist. Dies kann während der Umstellung zu einigen Unannehmlichkeiten führen, da bereits vorhandene Ergebnisse auf ältere Versionen bezogen werden, aber langfristig zahlt es sich aus.
Außerdem spielt die Art der Sequenzierungsdaten eine Rolle. Reads, die aus DNA-seq-, ChIP-seq- oder Hi-C-Protokollen stammen, werden an der Genomreferenzsequenz ausgerichtet. Da jedoch die von der DNA transkribierte RNA zu mRNA weiterverarbeitet wird (d. h. Introns werden entfernt), können viele RNA-seq-Reads nicht an einer Genomreferenzsequenz ausgerichtet werden. Stattdessen müssen wir sie entweder an Transkriptom-Referenzsequenzen ausrichten oder split-aware Aligner verwenden (siehe unten), wenn wir die Genomsequenz als Referenz verwenden. Damit verbunden ist die Wahl der Quelle für die Annotation der Referenzsequenz, d. h. der Datenbank mit den Koordinaten der Gene, Transkripte, Zentromere usw. Ich verwende normalerweise die GENCODE-Anmerkung da es eine umfassende Genannotation und Transkriptsequenzen kombiniert.
Eine lange Liste von Short-Read-Sequence-Alignment-Tools wurde entwickelt (siehe den Abschnitt Short-Read-Sequence-Alignment hier). Reviewing them is beyond the scope of this article (details about the algorithms behind these tools can be found hier). Meiner Erfahrung nach sind die beliebtesten Bowtie2, BWA, HISAT2, Minimap2, STAR und TopHat. Ich empfehle Ihnen, Ihr Aligner unter Berücksichtigung von Schlüsselfaktoren wie der Art der HTS-Daten auszuwählen und Anwendung sowie die Akzeptanz in der Gemeinschaft, die Qualität der Dokumentation und die Anzahl der Nutzer. Man braucht z. B. Aligner wie STAR oder Bowtie2, die Exon-Exon-Verbindungen bei der Zuordnung von RNA-seq zum Genom berücksichtigen.
Den meisten Mappern ist gemeinsam, dass sie die als Referenz verwendete Sequenz indizieren müssen, bevor das eigentliche Alignment stattfindet. Dieser Schritt kann zeitaufwendig sein, muss aber nur einmal für jede Referenzsequenz durchgeführt werden. Die meisten Mapper speichern die Alignments in SAM/BAM-Dateien, die nach dem SAM/BAM-Format (BAM-Dateien sind binäre Versionen von SAM-Dateien). Das Alignment gehört zu den rechenintensivsten und zeitaufwändigsten Schritten bei der Analyse von Sequenzierungsdaten, und SAM/BAM-Dateien sind sehr groß (in der Größenordnung von Gigabytes). Daher müssen Sie sicherstellen, dass Sie über die erforderlichen Ressourcen verfügen (siehe den letzten Abschnitt unten), um das Alignment in einer angemessenen Zeit durchzuführen und die Ergebnisse zu speichern. Aufgrund der Größe und des Binärformats von BAM-Dateien sollten Sie sie nicht mit Texteditoren öffnen, sondern Unix-Befehle oder spezielle Tools wie SAMtools.
Von den Alignments
Ich würde sagen, dass es nach dem Abgleich keinen eindeutigen gemeinsamen Schritt gibt, da sich an diesem Punkt jeder HTS-Datentyp und jede Anwendung unterscheiden kann.
Eine gängige nachgelagerte Analyse für DNA-seq-Daten ist das Varianten-Calling, d. h. die Identifizierung von Positionen im Genom, die im Vergleich zur Genomreferenz und zwischen Individuen variieren. Ein beliebter Analyserahmen für diese Anwendung ist GATK für Einzelnukleotid-Polymorphismus (SNP) oder kleine Insertionen/Deletionen (Indels) (Abbildung 2). Varianten, die größere DNA-Blöcke umfassen (auch als Strukturvarianten bezeichnet), erfordern spezielle Aufrufmethoden (siehe dieser Artikel für einen umfassenden Vergleich). Wie bei den Alignern empfehle ich, bei der Auswahl des richtigen Tools Schlüsselfaktoren wie die Art der Varianten (SNP, Indel oder strukturelle Varianten), die Akzeptanz in der Gemeinschaft, die Qualität der Dokumentation und die Anzahl der Nutzer zu berücksichtigen.
Die wahrscheinlich häufigste Anwendung von RNA-seq ist die Quantifizierung der Genexpression. In der Vergangenheit mussten die Reads an die Referenzsequenz angeglichen werden, und dann wurde die Anzahl der Reads, die an ein bestimmtes Gen oder Transkript angeglichen wurden, als Näherungswert für die Quantifizierung seiner Expressionswerte verwendet. Dieser Ansatz von Alignment und Quantifizierung wird von Tools wie Manschettenknöpfe, RSEM oder featureCounts. Dieser Ansatz wird jedoch zunehmend von neueren Methoden überholt, die in Software wie Kallisto und Lachs. Mit solchen Werkzeugen muss nicht die gesamte Sequenz eines Read an die Referenzsequenz angeglichen werden. Stattdessen müssen wir nur genügend Nukleotide alignieren, um sicher zu sein, dass ein Read von einem bestimmten Transkript stammt. Vereinfacht ausgedrückt, wird der Ansatz von Alignment und Quantifizierung auf einen einzigen Schritt reduziert. Dieser Ansatz wird als Pseudo-Mapping bezeichnet und erhöht die Geschwindigkeit der Quantifizierung der Genexpression erheblich. Auf der anderen Seite ist zu beachten, dass Pseudo-Mapping nicht für Anwendungen geeignet ist, bei denen ein vollständiges Alignment erforderlich ist (z. B. Variantenaufruf aus RNA-seq-Daten).
Ein weiteres Beispiel für die Unterschiede in den nachgelagerten Analyseschritten und den erforderlichen Werkzeugen bei sequenzierungsbasierten Anwendungen ist ChIP-seq. Die mit dieser Technik erzeugten Reads werden für das Peak-Calling verwendet, das darin besteht, Regionen im Genom mit einem signifikanten Überschuss an Reads aufzuspüren, die anzeigen, wo das Zielprotein gebunden ist. Es gibt mehrere Peak-Caller und diese Veröffentlichung untersucht sie. Als letztes Beispiel möchte ich die Hi-C-Daten nennen, bei denen Alignments als Input für Tools verwendet werden, die die Interaktionsmatrizen und daraus die 3D-Merkmale des Genoms bestimmen. Es würde den Rahmen dieses Artikels sprengen, alle sequenzierungsbasierten Assays zu kommentieren (eine relativ vollständige Liste finden Sie unter dieser Artikel).
Bevor Sie anfangen...
Der verbleibende Teil dieses Artikels befasst sich mit Aspekten, die nicht unbedingt als Schritte bei der Analyse von HTS-Daten angesehen werden können und die weitgehend ignoriert werden. Im Gegensatz dazu behaupte ich, dass es von entscheidender Bedeutung ist, dass Sie über die Fragen nachdenken, die in Tabelle 1 bevor Sie mit der Analyse von HTS-Daten (oder überhaupt von Daten) beginnen, und ich habe über diese Themen geschrieben hier und hier.
Tabelle 1
| Überlegen Sie es sich | Vorgeschlagene Maßnahme |
| Verfügen Sie über alle für die Analyse erforderlichen Informationen zu Ihrer Probe? | Systematische Erfassung der Metadaten der Experimente |
| Werden Sie in der Lage sein, Ihre Probe eindeutig zu identifizieren? | Einrichtung eines Systems, das jeder Probe eine eindeutige Kennung zuweist |
| Wo werden die Daten und Ergebnisse gespeichert? | Strukturierte und hierarchische Organisation der Daten |
| Werden Sie in der Lage sein, mehrere Proben nahtlos zu verarbeiten? | Skalierbarkeit, Parallelisierung, automatische Konfiguration und Modularität des Codes |
| Können Sie oder jemand anderes die Ergebnisse reproduzieren? | Dokumentieren Sie Ihren Code und Ihre Verfahren! |
Wie bereits erwähnt, liegen die HTS-Rohdaten und einige der bei ihrer Analyse erzeugten Dateien in der Größenordnung von Gigabytes, so dass es nicht ungewöhnlich ist, dass ein Projekt mit Dutzenden von Proben Terabytes an Speicherplatz benötigt. Außerdem sind einige Schritte bei der Analyse von HTS-Daten rechenintensiv (z. B. das Alignment). Die für die Analyse von HTS-Daten erforderliche Speicher- und Datenverarbeitungsinfrastruktur ist jedoch ein wichtiger Aspekt, der oft übersehen oder nicht diskutiert wird. Als Beispiel haben wir im Rahmen einer kürzlich durchgeführten Analyse Dutzende von veröffentlichten Arbeiten durchgesehen, die phänomenweite Assoziationsanalysen (PheWAS) durchführen. Bei modernen PheWAS werden 100-1.000 genetische Varianten und Phänotypen analysiert, was zu einer erheblichen Datenspeicherung und Rechenleistung führt. Dennoch ging praktisch keine der von uns geprüften Arbeiten auf die für die PheWAS-Analyse erforderliche Infrastruktur ein. Es überrascht daher nicht, dass ich Ihnen empfehle, die Speicher- und Rechenanforderungen, mit denen Sie konfrontiert werden, im Voraus zu planen und sie mit der Gemeinschaft zu teilen.
Benötigen Sie Hilfe bei der Analyse von DNA-Sequenzierungsdaten? Nehmen Sie Kontakt auf mit freelance bioinformatics specialist und Genomik-Experten auf Kolabtree.
Kolabtree helps businesses worldwide hire freelance scientists and industry experts on demand. Our freelancers have helped companies publish research papers, develop products, analyze data, and more. It only takes a minute to tell us what you need done and get quotes from experts for free.


Unlock Corporate Benefits
• Secure Payment Assistance
• Onboarding Support
• Dedicated Account Manager
Sign up with your professional email to avail special advances offered against purchase orders, seamless multi-channel payments, and extended support for agreements.