

FDA-Einreichungen Berater und Verfasser von Rechtsvorschriften Samradni Patil bietet eine Checkliste für 510k-Einreichungen to help Medizinprodukt Unternehmen mit schneller und einfacher FDA-Zulassung.
Das 510(k)-Antragsverfahren wird in der Regel für Medizinprodukte der Klasse II verwendet, um die Genehmigung der US Food and Drug Administration (FDA) zu erhalten. Das Premarket Approval (PMA)-Verfahren wird in der Regel für Medizinprodukte der Klasse III angewandt.
Im Rahmen des 510(k)-Prüfverfahrens wird die wesentliche Gleichwertigkeit (Substantial Equivalence, SE) mit einem ähnlichen, rechtmäßig in Verkehr gebrachten Produkt, auch Prädikatprodukt genannt, festgestellt. Das Produkt muss mindestens so sicher und wirksam sein wie das rechtmäßig in Verkehr gebrachte Produkt, damit es als substanziell gleichwertig mit diesem gelten kann. Das Produkt, das einer 510(k)-Prüfung unterzogen wird, muss Folgendes nachweisen, um den Anspruch auf SE mit dem Prädikatsprodukt zu erheben:
- Gleicher Verwendungszweck wie das rechtmäßig in Verkehr gebrachte Produkt (Prädikat) Prädikat
- Die gleichen technischen Eigenschaften wie das Prädikatgerät oder
- Unterschiedliche technologische Merkmale und Informationen/Prüfungen, die darauf hindeuten, dass das Produkt ebenso sicher und wirksam ist wie das Prädikatsprodukt, und andere Fragen zur Sicherheit und Wirksamkeit als das Prädikatsprodukt werden nicht aufgeworfen.
Die Nichterfüllung der oben genannten Kriterien führt zur Feststellung der Nicht-Substanziellen Äquivalenz (NSE).
Checkliste für 510k-Einreichungen
Das 510(k)-Prüfungsverfahren der FDA lässt sich grob in 2 Schritte unterteilen.
- Akzeptanzprüfung
- Substanzielle Überprüfung
Überprüfung von Zeitplänen
| Überprüfung Typ | Zeitplan (Kalendertage) | Prozess Ergebnis |
| Akzeptanzprüfung | Bis Tag 15 | Die FDA teilt dem Antragsteller mit, ob der Antrag zur Substanzielle Überprüfung oder In RTA-Haltezustand versetzt |
| Substanzielle Überprüfung | Bis Tag 60 | Interaktive Überprüfung oder Zusätzliche Informationen anfordern |
Hinweis: Tag 1 ist der Tag, an dem die FDA den 510(k)-Antrag erhält.
Lassen Sie uns zunächst erörtern, mit welchen Problemen Medizinprodukteunternehmen bei diesen Prüfverfahren konfrontiert werden.
Akzeptanzprüfung
Wird das 510(k)-Produkt in dieser Phase nicht akzeptiert, wird es in die Annahmeverweigerung (RTA) Halten. Gemäß FDA-DatenIm Jahr 2018 wurden etwa 30% 510(k)-Anträge in die RTA-Haltephase versetzt.
Substanzielle Überprüfung
Das nachstehende Schaubild zeigt den Prozentsatz der von der FDA in der Phase der Sachprüfung gestellten Anträge auf zusätzliche Informationen.
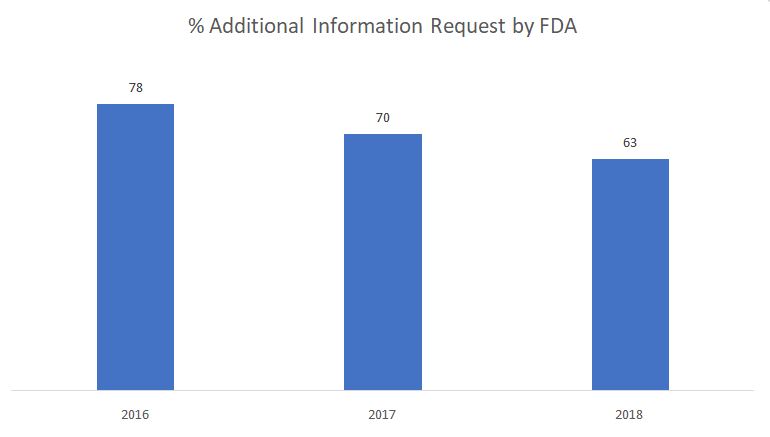

Quelle: FDA
Der Prozentsatz von 510ks, bestimmt als Nicht-substanziell gleichwertig (NSE) ist unten dargestellt.
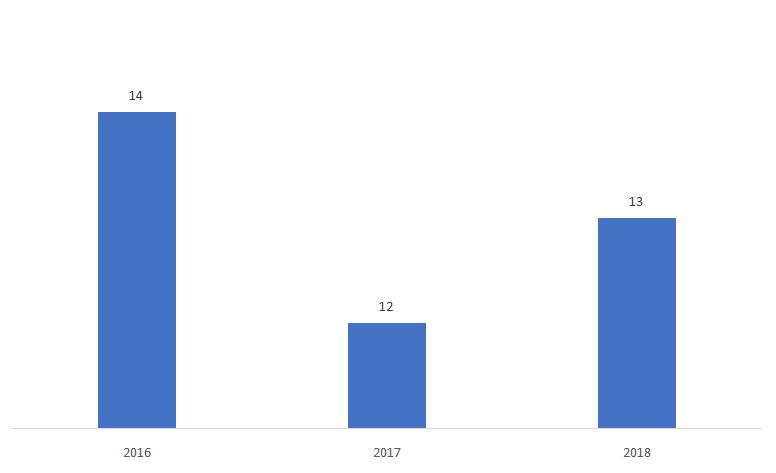

Quelle: FDA
Allgemeine Anforderungen an zusätzliche Informationen in der Checkliste für die 510k-Einreichung
Nachdem wir nun wissen, welche Fragen im Rahmen des 510k-Überprüfungsverfahrens häufig aufgeworfen werden, wollen wir uns nun darauf konzentrieren, welche Arten von Anfragen nach zusätzlichen Informationen üblich sind.
Die FDA-Daten stellt die folgenden Arten von Anfragen nach zusätzlichen Informationen dar:
- Unzureichende Gerätebeschreibung
- Unstimmigkeiten in der gesamten Einreichung - Unstimmigkeiten in dieser Kategorie beziehen sich meist auf die Beschreibung des Produkts oder die Anwendungshinweise
- Probleme mit Anwendungshinweisen
- Nichteinhaltung oder anderweitige Nichtbeachtung aktueller Leitfäden oder anerkannter Normen
- Die für bestimmte Gerätetypen erforderlichen Leistungstests fehlen völlig (d. h. es werden überhaupt keine Leistungsdaten bereitgestellt).
- Die für bestimmte Gerätetypen erforderlichen klinischen Daten fehlen vollständig (d. h. es werden überhaupt keine klinischen Daten geliefert).
Lassen Sie uns nun die besten Praktiken besprechen, die bei der Vorbereitung und Einreichung eines 510k-Antrags zu beachten sind.
Checkliste für die 510k-Einreichung:
1. Schreiben zur Annahmeverweigerung (RTA)
Der Zweck der Akzeptanzprüfung in der Anfangsphase besteht darin, zu prüfen, ob der 510k-Antrag verwaltungstechnisch vollständig ist. Es wird dringend empfohlen, die Checkliste für die Annahme im Leitfaden "Politik der Annahmeverweigerung für 510ks".
Um eine erfolgreiche Abnahmeprüfung zu gewährleisten, wird jedem Unternehmen empfohlen, die Elemente der folgenden Tabelle aus dem Leitfaden zu befolgen.
- Tabelle der vorläufigen Fragen: Obwohl diese Checkliste für den leitenden Prüfer gedacht ist, um eine erste Entscheidung zu treffen, ist es sehr empfehlenswert, diese Fragen informell zu beantworten, bevor der Antrag bei der FDA eingereicht wird.
- Tabelle "Organisatorische Elemente": Diese Elemente helfen dabei, die 510(k)-Anmeldung so zu organisieren, dass die Informationen in der 510(k)-Anmeldung leicht identifiziert werden können.
- Elemente einer vollständigen Einreichung (RTA-Positionen) Tabelle: Die Unternehmen sollten den in dieser Tabelle aufgeführten Elementen größere Aufmerksamkeit schenken, da diese Elemente entscheidend dafür sind, dass sie kein RTA-Schreiben erhalten.
2. Unzureichende Gerätebeschreibung
Die Gerätebeschreibung ist im 510(k)-Antrag obligatorisch. Es wird empfohlen, in diesem Abschnitt eine kurze Beschreibung und technische Spezifikationen hinzuzufügen. Alle Modelle und Zubehörteile des Medizinprodukts sollten enthalten sein. Bilder, Diagramme, Abmessungen, Zeichnungen und Toleranzen für jede Komponente sollten enthalten sein. Das Fehlen eines wichtigen Modells oder Zubehörs kann zu Verwirrung und zusätzlichen Fragen führen. Ungeeignete technische Spezifikationen können zu Missverständnissen und der Forderung nach zusätzlichen Tests führen.
3. Widersprüchliche Informationen in der gesamten Einreichung
- Inkonsistenz in der Gerätebeschreibung: Wenn ein Unternehmen beschließt, einen 510(k)-Antrag zu stellen, um ein zusätzliches Modell hinzuzufügen, ist es wichtig, dass die entsprechenden Abschnitte wie Anschreiben, Gerätebeschreibung, Kennzeichnung, Diskussion über die substanzielle Äquivalenz und leistungsbezogene Abschnitte mit der tatsächlichen Änderung in Einklang gebracht werden. Unstimmigkeiten können zu administrativen Verzögerungen und im schlimmsten Fall zur Anforderung zusätzlicher Tests führen.
- Unstimmigkeiten in der Indikation für den Gebrauch: Ähnlich wie bei der Beschreibung des Geräts können Unstimmigkeiten bei der Gebrauchsanweisung in verschiedenen Abschnitten der 510(k) Probleme verursachen. Die Angabe der Gebrauchsanweisung ist sehr wichtig, um eine SE-Bestimmung vorzunehmen.
Unstimmigkeiten in verschiedenen Abschnitten könnten durch eine sorgfältige Überprüfung des Antrags vor der Einreichung bei der FDA leicht vermieden werden. Es ist immer eine gute Idee, einen zusätzlichen Blick auf verschiedene Abschnitte zu werfen, um solche Fehler zu vermeiden.
4. Anderer Verwendungszweck als der des Prädikatsgeräts
Um eine SE-Bestimmung von der FDA zu erhalten, muss das Gerät folgende Eigenschaften aufweisen dieselbe Zweckbestimmung wie das Prädikatsprodukt. Dies ist wichtig, weil ein anderer Verwendungszweck als der des Prädikatsprodukts zu anderen Fragen der Sicherheit und Wirksamkeit führen kann. In einem solchen Fall ist 510(k) möglicherweise kein geeigneter Weg, um die Produktzulassung zu erhalten. Es ist zu beachten, dass Unterschiede in der Anwendungsindikation zwischen dem Prädikatsprodukt und dem Produkt, das einer 510(k)-Prüfung unterzogen wird, nicht unbedingt zu einer unterschiedlichen Zweckbestimmung führen müssen. Die Unternehmen sollten zusätzliche Anstrengungen unternehmen, um eindeutig nachzuweisen, dass die Unterschiede in der Anwendungsindikation nicht zu einer unterschiedlichen Zweckbestimmung geführt haben.
Der FDA-Leitfaden "Das 510(k)-Programm: Bewertung der wesentlichen Gleichwertigkeit bei Anmeldungen vor dem Inverkehrbringen [510(k)]"sollten herangezogen werden, um Klarheit über die mit der beabsichtigten Verwendung verbundenen Fragen zu erhalten.
5. Unzureichende/fehlende Testinformationen
- Unzureichende Testinformationen:
Es ist wichtig, die für ein bestimmtes Produkt geltenden Prüfungen zu kennen. Je nach Art des Produkts können Prüfungen der elektrischen Sicherheit, der elektromagnetischen Verträglichkeit (EMV), der Biokompatibilität, der Softwarevalidierung, der Sterilisation und der Benutzerfreundlichkeit erforderlich sein, um die Sicherheit und Wirksamkeit eines Produkts zu gewährleisten.
Oftmals unterschätzen Unternehmen den Umfang der erforderlichen Tests oder versuchen zu begründen, warum sie bestimmte Tests nicht durchführen.
Beispiel: Unternehmen können sich auf die Biokompatibilitätsdaten eines ähnlichen Produkts stützen, um die Biokompatibilität ihres Produkts zu behaupten. Dieser Ansatz kann in einigen Fällen akzeptabel sein. In vielen Fällen kann jedoch der Herstellungsprozess zwischen diesen Produkten eine separate Biokompatibilitätsprüfung des Produkts im Rahmen der 510(k)-Prüfung rechtfertigen.
Solche zusätzlichen Tests können mehrere Wochen dauern. Wenn die FDA die Durchführung dieser zusätzlichen Tests während der 510(k)-Prüfung verlangt, verlängert sich die Zeit bis zur endgültigen 510(k)-Zulassung erheblich.
Appropriate teams should give thorough consideration to current FDA guidance, product design, risk management process to make determination about amount of testing required.
- Fehlende Testinformationen:
Es ist wichtig, den Unterschied zwischen der traditionellen 510(k) und der speziellen 510(k) zu verstehen. Alle Testdaten müssen in den traditionellen 510(k)-Antrag aufgenommen werden.
6. Nichteinhaltung oder anderweitige Nichtbeachtung Aktuell Leitfaden(e) oder anerkannte Normen
Wie bereits erwähnt, muss sichergestellt werden, dass die Prüfungen so durchgeführt werden, dass die Einhaltung der neuesten anerkannten Normen nachgewiesen wird. Die neueste Version der Norm kann erhebliche Änderungen im Vergleich zur vorherigen Version aufweisen. Dies kann zusätzliche Fragen zur Sicherheit und Wirksamkeit aufwerfen, wenn das Produkt nicht nach der neuesten Version der Norm geprüft wird.
Die Bezugnahme auf die neuesten Leitliniendokumente hilft dabei, die Erwartungen oder Empfehlungen der FDA zu verstehen. Dies erleichtert auch den Überprüfungsprozess der FDA, da die Einreichung in einem für den Prüfer leicht verständlichen Format verfasst ist.
7. NSE-Bestimmung
Das endgültige Ziel des 510(k)-Verfahrens ist die Feststellung der wesentlichen Gleichwertigkeit (Substantial Equivalence, SE) mit einem Prädikatsprodukt. Wenn die FDA in der Phase der inhaltlichen Prüfung zusätzliche Informationen anfordert, sollten die Unternehmen jede Anforderung sorgfältig prüfen und wissenschaftlich fundierte Antworten geben. Werden die angeforderten Daten/Antworten nicht vorgelegt, kann dies zu einer NSE-Bestimmung führen. Viele NSE-Entscheidungen sind auf die fehlende Bereitstellung von Leistungsdaten zurückzuführen.
Es wird dringend empfohlen, die FDA zu konsultieren und mit ihr zusammenzuarbeiten, um die Erwartungen in dieser Phase zu verstehen. Hier sind einige weitere häufige Fehler, die mir in meiner Erfahrung aufgefallen sind.
Administrative Aspekte
8. Einreichung des Antrags an die richtige Adresse
Dies ist ein leicht vermeidbares menschliches Versagen. Informieren Sie sich immer auf der FDA-Website über die richtige Adresse um Ihre Bewerbung einzureichen.
9. Einschließlich gedruckter und elektronischer Kopien gemäß der neuesten FDA-Empfehlung
Die FDA stellt spezifische Anforderungen an die Anzahl der Kopien und elektronischen Kopien, die für einen 510(k)-Antrag eingereicht werden müssen. Derzeit ist 1 Papierkopie und 1 elektronische Kopie erforderlich. Stellen Sie keine Vermutungen an. Informieren Sie sich auf der Website der FDA, bevor Sie voreilige Schlüsse ziehen.
10. eCopy-bezogene Probleme
Die Einreichung muss den technischen Standards von eCopy entsprechen. Siehe dazu die eCopy-Anleitung um eine eCopy des Antrags vorzubereiten.
Die PDF-Namenskonvention und die empfohlene Dateigröße müssen eingehalten werden, um eCopy Hold Letter zu vermeiden. Auch wenn die Verwendung des eSubmitter-eCopies Werkzeug freiwillig ist, hilft dieses Tool bei der Validierung von eCopy gemäß den Empfehlungen der FDA.
11. Übermittlung von Informationen an den Gutachter
Nach Erhalt des ersten Hold-Letters der FDA (RTA-Hold, eCopy-Hold) machen Unternehmen oft einen Fehler bei der Übermittlung von Informationen an den Prüfer. Prüfen Sie die von der FDA erhaltene E-Mail oder die entsprechende Anleitung der FDA, wohin die Antwort auf das Hold-Letter zu senden ist.
Technische Aspekte
12. Traditionelle 510(k) vs. spezielle 510(k)
Unternehmen können den Fehler machen, Anträge als traditionelle 510(k) oder spezielle 510(k) zu kategorisieren. Der Hauptunterschied zwischen der traditionellen 510(k) und der speziellen 510(k) ist die Zeit, die für die Prüfung des Antrags durch die FDA erforderlich ist. Special 510(k) dauert 30 Kalendertage, während Traditional 510(k) 90 Kalendertage in Anspruch nimmt. Ich habe mehrfach erlebt, dass die FDA Unternehmen aufgefordert hat, eine spezielle 510(k) in eine traditionelle 510(k) umzuwandeln. In diesem Fall verschwenden die Unternehmen so viel Zeit mit dem Umwandlungsprozess.
Gemäß der FDA kann eine spezielle 510(k)-Zulassung angebracht sein, wenn:
- Der Änderungsvorschlag wird von dem Hersteller eingereicht, der rechtlich befugt ist, das bestehende Produkt in Verkehr zu bringen;
- Leistungsdaten sind nicht erforderlich, bzw. wenn Leistungsdaten erforderlich sind, stehen bewährte Methoden zur Bewertung der Änderung zur Verfügung; und
- Alle Leistungsdaten, die zum Nachweis der wesentlichen Gleichwertigkeit erforderlich sind, können in Form einer Zusammenfassung oder einer Risikoanalyse überprüft werden.
To avoid such mistake, do thorough Forschung on the FDA database to identify if similar change was submitted as Special 510(k) or Traditional 510(k). Refer to the Leitfäden von der FDA. Wenn Sie immer noch Zweifel haben, nehmen Sie die Hilfe von Regulierungsberatern in Anspruch. Verwenden Sie einen risikobasierten Ansatz. Wenn Sie immer noch Zweifel haben, empfiehlt es sich, einen konservativen Ansatz zu wählen und ein traditionelles 510(k) einzureichen.
Einzigartige Herausforderungen
13. Art der Einrichtung
Einige Geräte können aufgrund ihrer besonderen Beschaffenheit besondere Fragen aufwerfen. Bestimmte technologische Bereiche wie Künstliche Intelligenz (KI) und Cybersecurity sind relativ neu. Die FDA hat mit der Industrie zusammengearbeitet, um Leitlinien für diese Bereiche zu entwickeln.
In solchen Fällen wird eine vorherige Besprechung mit der FDA vor der 510(k)-Einreichung dringend empfohlen. Verweisen Sie die FDA-Leitfaden wenn Unternehmen vor der 510(k)-Einreichung ein Feedback und ein Treffen mit der FDA benötigen.
Schlussfolgerung
Dies waren die wichtigen Punkte einer Checkliste für die 510(k)-Einreichung. Die RTA-Briefe (Refuse to Accept) der FDA ließen sich durch eine sorgfältige Prüfung des Antrags und die Befolgung der FDA-Leitlinien leicht vermeiden.
Zusätzliche Informationen, die im Rahmen des inhaltlichen Überprüfungsverfahrens angefordert werden, könnten durch das Verfassen klarer und prägnanter 510(k)-Anträge reduziert werden. Dies ist oft eine Kombination aus Wissenschaft, Kunst und Erfahrung. Es wird dringend empfohlen, die Hilfe von erfahrenen Berater für regulatorische Angelegenheiten bei der Erstellung von 510(k)-Zulassungsanträgen, um kostspielige Fehler zu vermeiden, befolgen Sie diese Checkliste für 510(k)-Anträge. Bleiben Sie in Bezug auf die geltenden Vorschriften, Normen und Leitfäden auf dem Laufenden, um die Chancen auf eine erfolgreiche Einreichung zu erhöhen.
Notwendigkeit der Konsultation eines Experte für FDA-Einreichungen? Zusammenarbeit mit erfahrenen Verfassern von Vorschriften, Experten der Medizinproduktebranche und 510k-Berater die Medizintechnikunternehmen bei der Erstellung von Zulassungsunterlagen für eine erfolgreiche FDA-Zulassung unterstützt haben.
Kolabtree helps businesses worldwide hire freelance scientists and industry experts on demand. Our freelancers have helped companies publish research papers, develop products, analyze data, and more. It only takes a minute to tell us what you need done and get quotes from experts for free.


Unlock Corporate Benefits
• Secure Payment Assistance
• Onboarding Support
• Dedicated Account Manager
Sign up with your professional email to avail special advances offered against purchase orders, seamless multi-channel payments, and extended support for agreements.